10x Genomics 在分析软件这一块还是非常给力的,开发的每个软件都非常好用而且操作简单,特别是安装方便,基本解压后就能用。对于 10x 的空间转录组数据主要用 10x 自己开发的 Space Ranger 来进行前期的处理。
软件下载地址
https://support.10xgenomics.com/spatial-gene-expression/software/downloads/latest
可以直接点击下载,也可以用下面的 curl 或 wget 命令下载。
基因组下载
在前面同一个页面往下拉就能看到。
10x 提供了专门的用于空间转录组分析的基因组,目前较新的基因组版本是 2020 年 6 月更新的,基因注释为 Ensembl 98 版本,主要包括人和小鼠。
系统要求
Linux 系统上运行底限要求:
8 核 Intel 或 AMD 处理器(建议使用 32 核)
64GB RAM(建议 128GB)
1TB 可用磁盘空间
64 位 CentOS / RedHat 6.0 或 Ubuntu 12.04
集群模式下运行需要额外满足以下要求:
每个节点 8 核 Intel 或 AMD 处理器
每个内核 6GB RAM
共享文件系统(例如 NFS)
SGE 或 LSF 批处理系统
软件安装
1,解压 SpaceRanger 文件
1 tar -xzvf spaceranger-1.1.0.tar.gz
2,解压参考基因组文件
1 tar -xzvf refdata-gex-GRCh38-2020-A.tar.gz
3,加入环境变量
1 export PATH=/opt/spaceranger-1.1.0:$PATH
输入文件准备
Fastq 序列文件
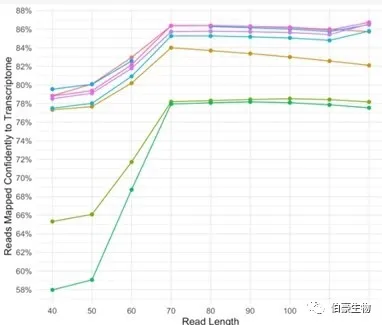
10x 建议测序 read2 设置 90 个碱基的长度 。 从下面 10x 测试的 6 个样本来看,reads 序列长度达到 70bp 以后,mapping 率就不太变化了,有些样本在长度大于 90bp 以后 mapping 率反而下降了。10x 给出这样的建议是考虑到建库后插入片段太短的话,太长的序列里面会包含部分 polyA 甚至 UMI 序列反而影响比对效果。 因为国内目前基本上是测 2x150bp 的,如果建库片段过短比对率较低的话建议将 read2 剪切成 90bp 来分析效果应该会好一点。
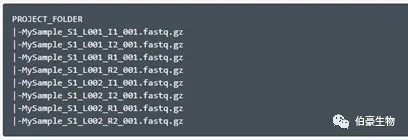
Fastq 文件名建议是如下格式的,以免运行报错。
图像文件:
Space Ranger 支持两种样式的显微镜图像:用苏木精和曙红(H&E)染色的明场图像,必须是 24 位彩色 TIFF,16 位灰度 TIFF 或 JPEG。Space Ranger 要求任一方向上的图像至少为 2000 像素。
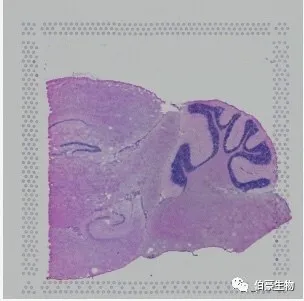
注意:图像边框的四个角位置是固定的,比如说 三角形在左下角 ,如果扫描出来的图片四个角的位置变了需要先手动调整过来再使用。
软件运行
一般我们拿到的都是直接是 fastq 序列文件了,所以可以直接跑 spaceranger count,不需要再去运行 spaceranger mkfastq 了。
spaceranger count 运行时图片对其有两种方式,一种软件自动识别图片进行对齐,另外一种就是先用 Loupe 软件手动对齐,生成对于 json 文件提供给后面的软件,后期会介绍怎么来手动调整图片。
自动对齐
1 $ cd /home/jdoe/runs
2 $ spaceranger count --id=sample345 \
3 --transcriptome=/opt/refdata/GRCh38-3.0.0 \
4 --fastqs=/home/jdoe/runs/HAWT7ADXX/outs/fastq_path \
5 --sample=mysample \
6 --image=/home/jdoe/runs/images/sample345.tif \ 7
7 --slide=V19J01-123 \
8 --area=A1
手动对齐
1 $ cd /home/jdoe/runs
2 $ spaceranger count --id=sample345 \
3 --transcriptome=/opt/refdata/GRCh38-3.0.0 \
4 --fastqs=/home/jdoe/runs/HAWT7ADXX/outs/fastq_path \
5 --sample=mysample \
6 --image=/home/jdoe/runs/images/sample345.tif \
7 --slide=V19J01-123 \
8 --area=A1 \
9 --loupe-alignment=sample345.json
参数说明:
--Id:结果输出文件夹名称
--Transcriptome:基因组目录
--Fastqs:fastq 文件木兰路
--Sample:原始样本名
--Image:镜像图片文件
--Slide:使用的 10x 芯片型号
--area:样本所在芯片的区域(四通道芯片位置从上到下分别为 A1、B1、C1、D1)
--loupe-alignmen: 图片手动对齐生成的 json 文件,如果是自动对齐不要需要此参数
另外,spaceranger 默认使用系统上可用的所有内核来执行管。可以使用 --localcores 选项来限制使用内核的数量。同理,可以使用 -localmem 来限制使用内存的大小。
结果输出:
成功运行后会到的下面的信息(包括主要的输出文件)
1 2016-11-10 16:10:09 [runtime] (join_complete) ID.sample345.SPATIAL_RNA_COUNTER_CS.SPATIAL_RNA_COUNTER_CS.SUMMARIZE_REPORTS 2
2
3 Outputs:
4 - Run summary HTML: /opt/sample345/outs/web_summary.html
5 - Outputs of spatial pipeline: /opt/sample345/outs/spatial
6 - Run summary CSV: /opt/sample345/outs/metrics_summary.csv
7 - BAM: /opt/sample345/outs/possorted_genome_bam.bam
8 - BAM index: /opt/sample345/outs/possorted_genome_bam.bam.bai
9 - Filtered feature-barcode matrices MEX: /opt/sample345/outs/filtered_feature_bc_matrix
10 - Filtered feature-barcode matrices HDF5: /opt/sample345/outs/filtered_feature_bc_matrix.h5
11 - Unfiltered feature-barcode matrices MEX: /opt/sample345/outs/raw_feature_bc_matrix
12 - Unfiltered feature-barcode matrices HDF5: /opt/sample345/outs/raw_feature_bc_matrix.h5
13 - Secondary analysis output CSV: /opt/sample345/outs/analysis
14 - Per-molecule read information: /opt/sample345/outs/molecule_info.h5
15 - Loupe Browser file: /opt/sample345/outs/cloupe.cloupe
16
17 Pipestance completed successfully!
空间转录组测序服务优势
高质量切片: 切片、贴片经验丰富,针对不同组织优化了解决方案。
流程化分析: 完善的分析流程,准确快速解析空间转录组数据。
标准化内控: 丰富的实操经验构建了标准化的内控体系。
专业化团队: 资深的技术团队具有多年项目方案设计、实验操作、售后分析等经验。
全流程服务: 提供组织冷冻包埋、贴片、切片、透化、建库测序及数据分析的全套服务。
更多伯豪生物人工服务:

