 |  |

单细胞测序技术为基础科研、临床诊断、药物研发等领域提供了诸多全新发现视角。现阶段主流的单细胞测序,大多是基于 10X Genomics、BD Rhapsody 等单细胞捕获设备获得 cDNA 后进行打断、扩增、建库,并用二代测序分析基因的整体定量。然而,基因在不同组织、不同细胞亚群中会使用 mRNA 的不同转录本,同时也包括 InCRNA;此外 SNV、融合基因等结构变异也具有组织和细胞特异性,目前基于二代测序的单细胞数据局限于 3'或 5' 端的 100~150bp,因此较难满足这类需求:而传统的 Smart-seq 虽然可以实现全长转录本覆盖,但转录本结构分析需要经过组装且细胞通量较低,成本较高,研究单细胞水平的可变剪切仍然较为困难。
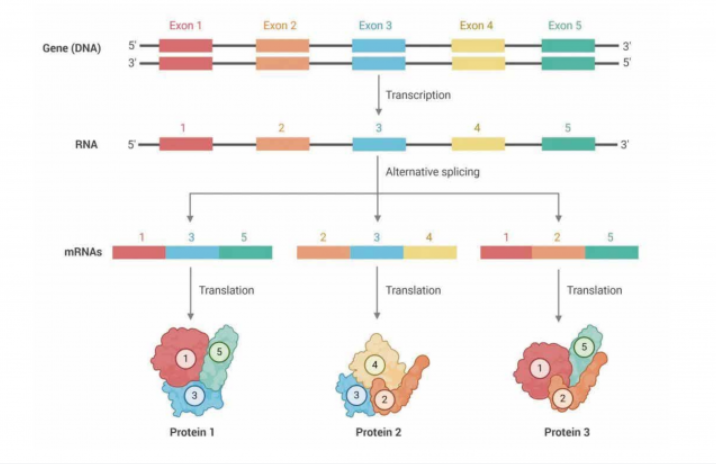
三代测序如 Pacbio、Nanopore 等技术能够以其长读长的优势解决这一痛点。因此,如果能将二代测序与三代测序相结合,既能获得 mRNA 的全长序列,并通过 Cell Barcode 信息定位到细胞亚群,即可解决这一单细胞研究领域的痛点。但是,我们在前期测试中发现,二代单细胞测序一般获得约 3 万个基因的表达矩阵,三代全长测序能获得超过 10 万个转录本的表达矩阵,两套数据的聚类图谱差异巨大,现有的分析流程并未很好的解决两套数据的整合问题。因此,如何从庞大的二代 + 三代,也即基因 + 转录本的单细胞数据中,挖掘到有价值的特异性转录本,能够为单细胞临床转化、药物靶点发现带来更加精细的挖掘角度。
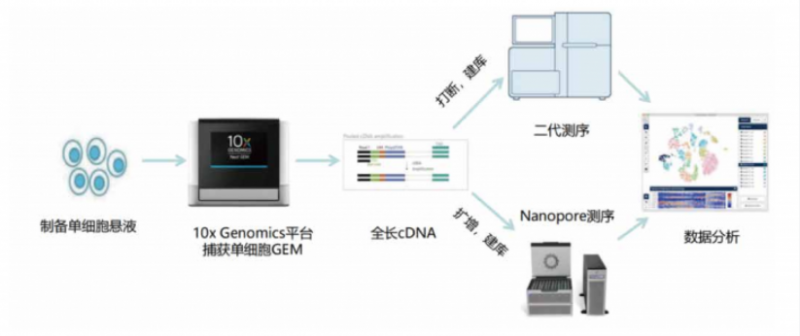
伯豪生物基于十多年的单细胞组学服务经验,可提供从样品保存、运输、单细胞悬液制备,到单细胞分选、建库和数据分析的解决方案。同时,及智医学团队出身单细胞科研服务行业,重点围绕单细胞富集与检测平台、单细胞测序技术平台和基于 AI 算法的单细胞数据分析算法平台。建立了单细胞转录组、空间转录组、单细胞联合 Bulk 多组学等多种独特的分析流程和方法,尤其擅长各类免疫细胞与基质细胞的分类、功能解析、细胞互作、药物靶点筛选等分析项目。最终通过积累的上百种单细胞分析方法与百万级别单细胞数据库,为单细胞临床转化类项目提供专业研发服务。团队生信专家通过高效的自动化分析脚本,并历时数月的二代 + 三代单细胞算法测试,目前已经解决了二代 + 三代单细胞聚类的诸多分析难点。
伯豪生物与及智医学强强联合,正式推出单细胞全长转录本测序服务,即单细胞 cDNA 水平的转录、遗传变异研究,通过一次捕获,两次建库,同时获得单细胞聚类与转录本信息:
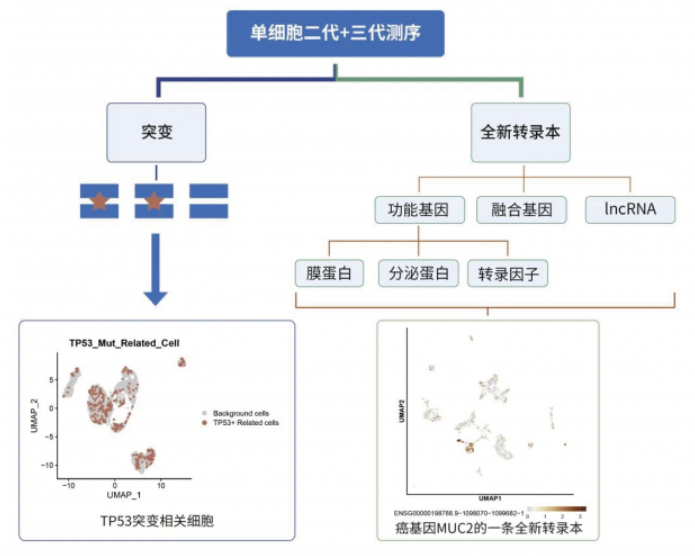
目前,该技术方向为如下科研问题,提供了潜在的解决办法:
1、发现不同细胞携带的突变,携带突变的细胞与非突变细胞相比、携带不同突变类型的细胞相比,挖掘基因表达规律;
2、挖掘功能基因,如膜蛋白、分泌蛋白、转录因子等编码基因的转录本使用情况,并发现全新功能的转录本;
3、发现融合基因所在的细胞亚群,研究它们与其它肿瘤细胞的拟时序分化关系;
4、发现亚群特异性的全新 IncRNA;
5、获得亚群特异性表达的转录本,能够辅助小核酸类药物开发企业,针对该特异性转录本设计 siRNA 干扰片段,提升小核酸干扰靶点的有效性。
案例解析
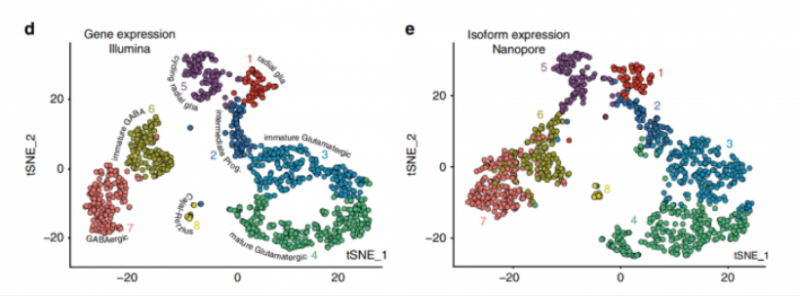
2021 年 11 月 11 日,来自澳大利亚 沃尔特 - 伊丽莎霍尔医学研究所的 Tian 等人开发了一种基于 Nanopore 测序和 10X Genomics 的全长转录组单细胞测序方法,分析单细胞中的全长异构体、可变剪接和突变检测。研究成果发表在国际知名期刊 GenomeBiology (IF=13.6),论文题目为 "Comprehensive characterization ofsingle-cell full-length isoforms in human and mouse with long-read sequencing"。
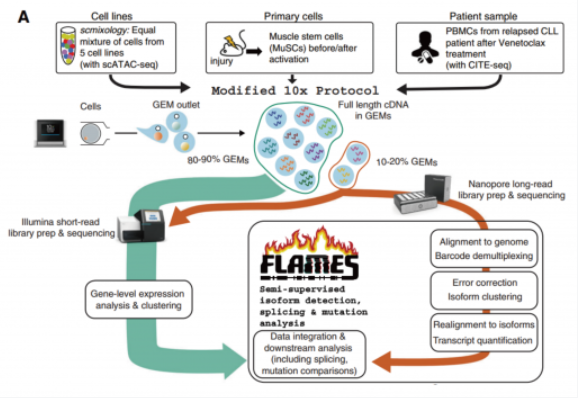
文章中,使用 10X Genomics 技术分选得到单细胞的全长 cDNA 后,将 cDNA 一分为二,一份进行打断建库用于二代测序,另一份进行全长扩增建库用于 Nanopore 三代测序。此时 Nanopore 的文库上也包含了细胞 Barcode, 后续可以通过分析流程将三代测序和二代测序结果通过细胞 Barcode 一 一 对 应。通过这样的方式,即实现了获得全长转录本,分析亚群的特征性转录本使用,并同时拿到了突变所在细胞。
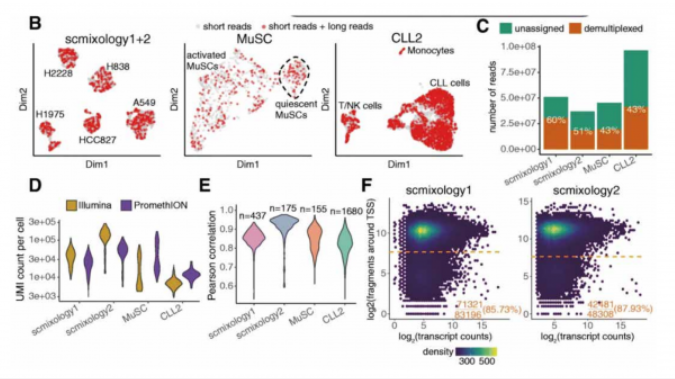
文章数据分析显示其中 40%-60% 的 Nanoporereads 可以分配给预期的 Barcode, 并保留用于后续分析(图 C)。在数据处理过程中,非全长且不能唯一分配给转录本的数据被丢弃。最终每个细胞的平均 UMI 为 10,000 至 60,000 个,并且与对应的短读数据情况相符(图 D)。Nanopore 和 Ilumina 数据的基因水平的 UMI 计数也高度一致(图 E)
文章数据分析显示其中 40%-60% 的 Nanoporereads 可以分配给预期的 Barcode, 并保留用于后续分析(图 C)。在数据处理过程中,非全长且不能唯一分配给转录本的数据被丢弃。最终每个细胞的平均 UMI 为 10,000 至 60,000 个,并且与对应的短读数据情况相符(图 D)。Nanopore 和 Ilumina 数据的基因水平的 UMI 计数也高度一致(图 E)

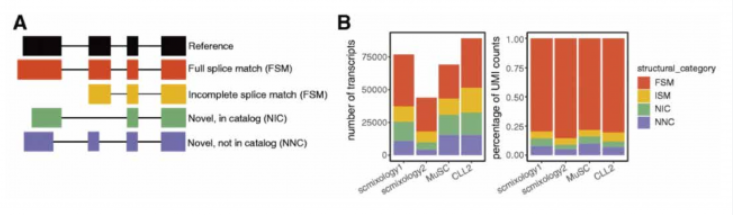
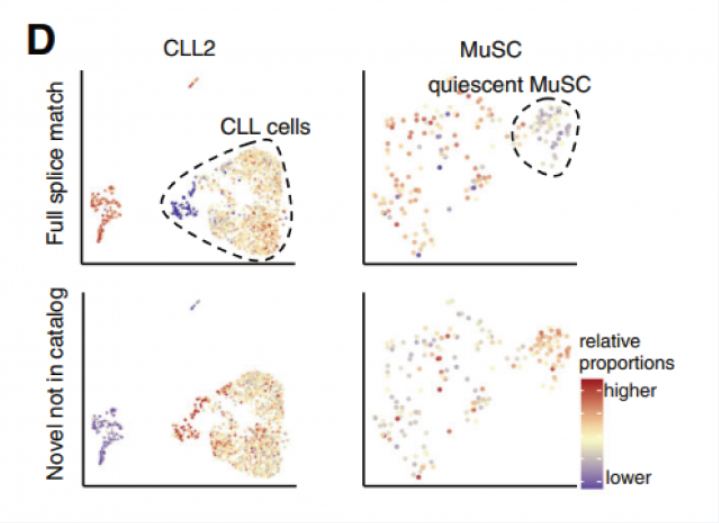
通过聚类分析发现,CLL(慢性淋巴细胞白血病)细胞相比正常免疫细胞具有更高比例的新型转录本,特别是新型剪接的转录本。同样,相比激活的干细胞,静态肌肉干细胞也有更高比例的新型转录本(图 D)。
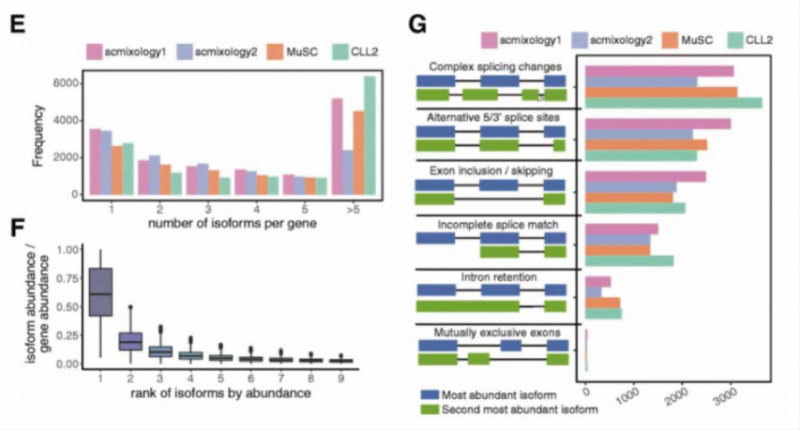
分析发现,约 80% 的基因可以表达多种转录本(图 E),但是大多数基因主要表达 1 到 2 种转录本类型(图 F),约 30% 的基因含有多于一种的可变剪接事件,意味着 2 个最高表达的异构体可能涉及多个外显子的复杂剪接变化而产生不同。
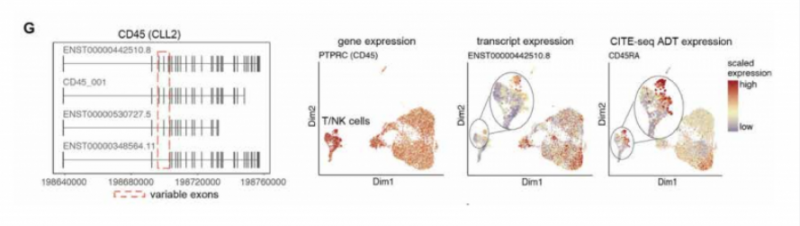
文章通过分析 CLL 数据,检测到 CD45 的多种亚型(图 G),CD45 的表达通过 CITE-seg 进行验证。CITEseg 可以同时检测 RNA 和细胞表面蛋白,这种方法结合三代测序,可以对细胞表面蛋白进行更深入的分析和探索。
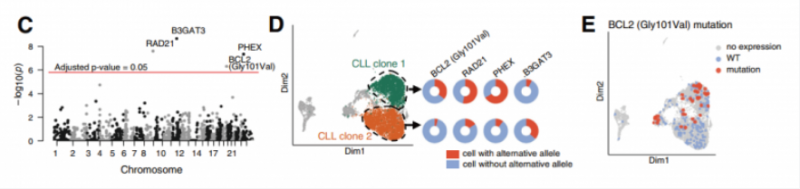
对 CLL 数据集进行分析,寻找只存在于癌细胞中的,且在不同的 CL 转录簇中具有不同等位基因频率的 SNVS,通过经典的曼哈顿图最终发现四个变异在不同的 CLL 聚类呈现显著差异(图 C, 图 D)。其中发现的 Gly101Val 突变,此突变已被证实通过降低 BCL2 对 venetoclax 的亲和力而使患者对 venetoclax 治疗产生耐药性,通过分析发现患者 CLL2 携带约 25% 的 GIV101Va| 突变,并发现该突变不仅属于亚克隆,而且与特定的转录簇相关(图 E)。
单细胞全长转录本测序的样品选择与实验细节
由于单细胞全长测序需要对 mRNA 反转录后的 cDNA 全长进行测序,核心是需要将完整的全长 cDNA 扩增至 2ug 的 Nanopore 建库起始量,而常规单细胞是将一链 cDNA 做基础扩增后全部打断用来做建库测序,因此,这一实验细节就意味着单细胞全长测序需要额外质控。
一、样品选择:
常规单细胞测序样品来源分为新鲜采集与液氮速冻两种类型,两种类型的样品需要两种处理方式,新鲜采集样品需要在 48h 内制备悬波并上机,液氮速冻样品需要将细胞膜破碎,丢弃细胞质,分离提取细胞核,用单个核来做单细胞测序。
不过,由于细胞核里面的 RNA 大多为初始 RNA,包含有较多内含子,而从初始 RNA 加工为成熱 mRNA 的过程大多发生在细胞质中,因此,抽核类的项目并不太适用于单细胞全长测序。虽然在 2022 年 7 月份一篇 Nature Biotechnoloey 的文章是对人脑抽核后的单细胞样品进行三代全长测序,不过由于拿不到成熟 mRNA,文章是站在了特定基因在不同亚群的外显子保留这样的科研角度统计规律(如下图)。文章角度非常新颖,也是科研界首次用单细胞全长测序发现,人脑中某些基因在不同亚群中使用不同的外显子组合,生成多种编码蛋白。不过,由于最终拿到的仍旧是细胞核内的 RNA,后续还需要大量验证工作,因此抽核后做单细胞全长测序的临床转化价值较小。所以,单细胞全长测序的项目最适宜采集新鲜样品制备细胞悬液,捕获成熟 mRNA 开展后续验证工作。
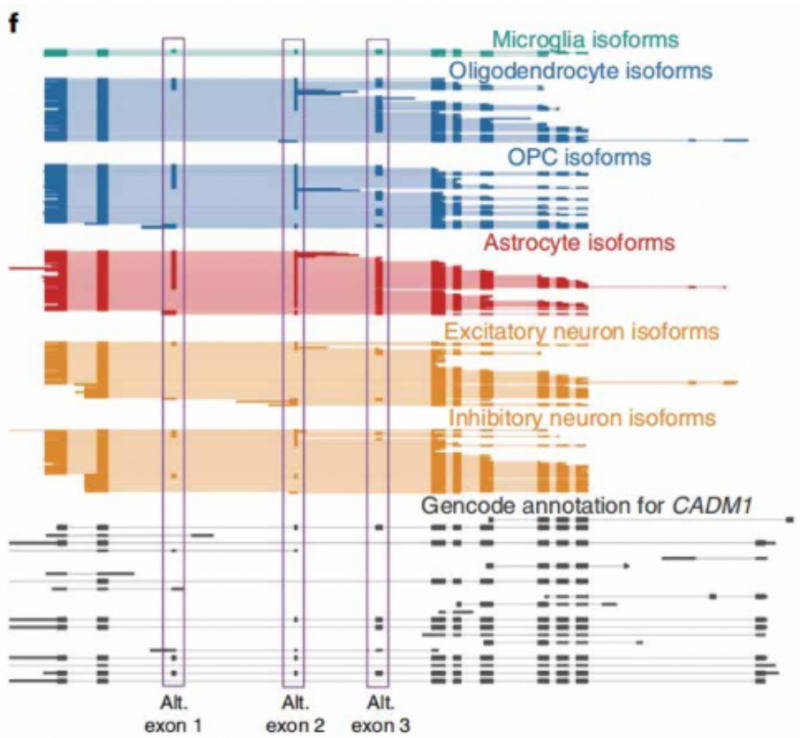
经三代单细胞全长测序发现 CADMI 基因在人脑神经元(兴奋性、抑制性)、星胶、小胶、少突细胞亚群中,会使用不同的外显子组合。原文也有用蛋白质谱技术对这些外显子的多肽产物进行验证的工作。
二、悬液质控:
在收集到新鲜样品之后,可以使用单细胞组织保护液(伯优®单细胞组织保存液)将样品在 24h-48h 内从临床运输至实验室进行悬液解离,并通过显微镜、细胞计数仪检测悬液质量。

由于全长单细胞对 RNA 质量要求较高,比较建议悬液活率在 85% 以上,同时用台盼蓝、AO/PI 双染鉴定,并用显微镜仔细观察细胞真实活率、红细胞比例(红细胞在光镜下,可以观察到圆饼状的亮圈,中间有黑色小点,有经验的单细胞实验员可以通过肉眼观察判断出来,而不少品牌的细胞计数仪有可能会把红细胞计算为碎片,甚至检测不到)。
另外,现阶段二代单细胞测序,单个样品的数据量大多为 100G,可以容纳 5000-8000 左右的细胞捕获量;而三代测序成本较高,站在节省经费的角度,建议一方面准确的对细胞悬液的浓度进行测定(不可单纯依靠细胞计数仪),来控制上机细胞总数(建议上机不超过 1 万个细胞);同时也要结合不同品牌单细胞捕获设备的真实捕获率(这点最好找成熟单细胞科研服务公司来完成)来进行综合判定(建议捕获不超 5000 个细胞如果超过 5000 需要增加三代测序数据量)。
三、文库制备
单细胞全长转录本测序,只需要一次捕获,拿到一链 CDNA 之后要立刻进行全长扩增,如下图:
因此,就需要将已扩增好的 cDNA 全长进行质控:

如上图,cDNA 条带主峰在 1 -1.5kb 左右,下一步可以联系三代测序工厂寄送样品,由他们进行建库测序。但是,也要测序工厂及时反馈三代文库的质检图片,要求文库主峰与 cDNA 条带主峰一致,方可进行正式的 Nanopore 上机测序实验。
四、单细胞测序剩余样本用于新的科研发现:
由于现阶段三代全长测序的准确性不够高,考虑到后续验证工作,比较建议在单细胞上机之后,将剩余的细胞样品进行冻存,从 DNA、RNA、蛋白三个层面开展后续验证实验:
1、DNA 水平:
在我们前期测试中发现,三代原始数据中基因单核苷酸结构变异 SNV(RNA 层面的 SNP、Indel) 较多,为了拿到准确的,与 DNA 层面一致的突变信息,就需要结合 DNA 层面的检测来共同筛选核心突变。有两种做法:
第一,同时将肿瘤患者的外周血和单细胞实验剩下的肿瘤细胞做全外显子测序(两个样品的市场价合计不超 5000 元),通过 肿瘤组织测出来的突变 扣掉 自身 PBMC 的胚系突变,可以得到体细胞突变,将这些突变 基因位点作为核心突变,利用自动化脚本,提取 三代数据中的原始 reads,这些 reads 都带有的 Cell barcode 信息可以定位到突变所在的细胞与亚群!即可通过拟时序算法分析突变细胞 vs 非突变细胞的发育分化轨迹。
第二:做全基因组重测序(可以根据具体课题决定是否还需收集 PBMC),发现拷贝数变异 CNV,以及融合基因信息,将这些信息与三代全长进行联合分析。后续分析内容也极为丰富,可以展开多个科研角度的解释。
2、RNA 水平:
在三代全长拿到特征性转录本之后,还需要做后续验证,如果序列较少,可以通过 5'RACE、3'RACE 实验拉全长获得准确序列;如果候选转录本序列较多,也可以通过 Pacbio 直接做 Bulk 测序(可以混样测一份即可,目的是拿到序列),再结合单细胞全长转录本的特异性表达规律,可以快速、低成本获得这些序列的完整信息,下一步即可通过构建动物模型,开展功能验证工作。
3、蛋白层面:
现阶段的单细胞测序大多是以基因作为靶点,但是从已经发表的上万篇单细胞数据中,也经常发现基因的表达特异性并不强,这个是现阶段单细胞测序需要升级改进的核心关键点。而在真实组织中,基因在不同亚群中使用不同的转录本编码多种蛋白产物。有了单细胞全长转录本技术,也就意味着可以将靶点发现从基因细化为转录本,挖掘转录本的蛋白编码产物。因此,临床转化最核心的一步:膜蛋白层面,可以依靠全长转录本获得一些全新的发现。
现有的蛋白质质谱技术无法做到 针对单个细胞进行广泛的蛋白质检测,但是蛋白质的编码序列都是从 RNA 层面的转录本翻译过来,转录本序列的检测比蛋白质的检测要容易很多。所以,这个里面就依托一套简单的逻辑:从 DNA 到 RNA 到蛋白的中心法则,即可做到通过单细胞全长转录本测序,发现亚群特异性转录本,再将转录本序列预测的多肽产物与蛋白质谱打出来的多肽产物进行匹配,发现一条潜在的转录本 + 编码产物,即为一条新型潜在靶点。其实,在肿瘤新抗原发现领域,这套序列预测 + 质谱检测的策略已经非常成熟并目较为实用,因此,可以基干中心法则将这套成熟策略转用到单细胞全长转录本发现新型蛋白编码产物领域。
总结
综上所述,单细胞全长转录本更适合做新鲜样品,整体实验过程并不复杂,基本上现阶段单细胞科技服务类公司都能实现,只需要在几个细节上稍加注意即可。
总结下来,单细胞全长测序的本质只是对转录本加了 细胞亚群 的标签,方便从数万条转录本快速筛选到特异性表达的少数转录本。这个并不是一套全新开发的技术,只能算是从 DNA 到 RNA 到蛋白的一整套符合中心法则的单细胞多组学的技术方案。在我们前期拜访前沿课题组的过程中,有不少研究员曾想过这样的方法,只是行业内缺乏前人尝试,我们深入思者过这些细节后,发现这套方案从样品的选择,测序实验,数据呈现,均比现阶段的单细胞二代测序更加实用,更加贴近临床转化。从另外一个角度,转录本是基因功能实现的最小细分单位,针对转录本研究的单细胞全长测序,算得上是转录组研究领域的终点站。
解析某肿瘤样本单细胞全长转录本的实测数据
一、基础质控
| 测序 | 二代测序 | 三代测序 |
| 测序数据量 | 100G | 100G |
| 总计测得 | 约 2 万条基因 | 44834 种转录本 |
| 中位值 | 基因中位值:2906 | 转录本中位值:1720 |
详细统计如下,实测基因是指在三代全长中测到的基因:
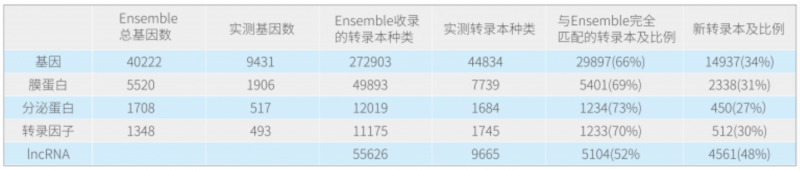
以膜蛋白为例,数据库中收录的人总膜蛋白有 5520 个,对应转录本有 49893 条;在该样品中测到了 1906 个(大部分膜蛋白不一定会在该肿瘤中表达),对应转录本是 7739,其中与 Ensemble 完全匹配的转录本有 5401 条,新转录本 2338 条,意味着平均每个膜蛋白会表达 1 条新转录本。从总体统计来看,功能基因(膜蛋白、分泌蛋白、转录因子)会约 30% 的新转录本,IncRNA 由于目前数据库收录的并不多,所以有约 50% 的新 IncRNA。
二、转录本表达的可靠性:
如同二代单细胞测序一样,三代全长测序同样会统计每条转录本在该样品中测到的总 count 数,截图如下:
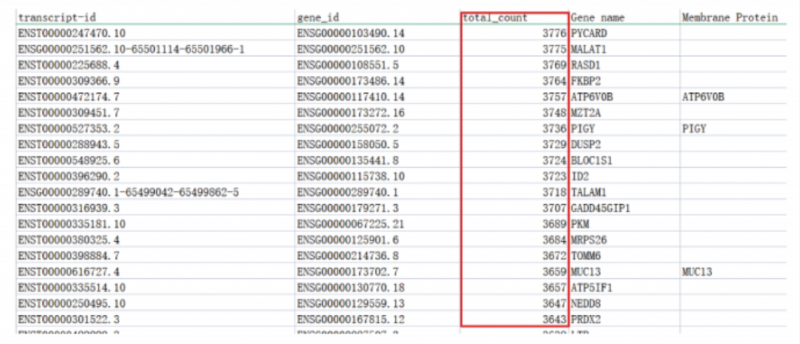
目前已经发表的 SCI 文章,转录本 total count>5 即可纳入正常聚类分析,经过实测建,议设置转录本 total count>20(约占总转录本的 80%),作为验证实验的可靠候选。
三、转录本表达的特异性
单细胞全长转录本测序的一大核心应用,是发现亚群的特征性转录本。单细胞二代 + 三代的合并聚类、注释如下:
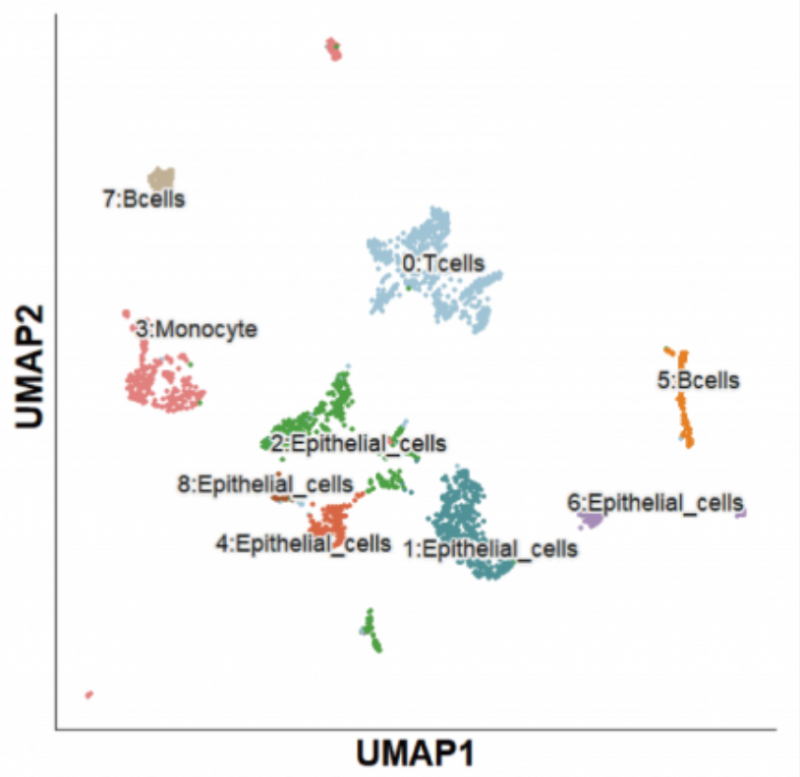
总计分 B cels、T cels、Monocyte cells、Epithelial cells 四大群。更精细的注释,如 Memory B、Treg、Naive T、ExhaustT、Mac 等我们会在后续章 节陆续展开讨论。
以 CART 治疗领域的某明星分子 MUC1 为例,二代测序基因表达情况如下如下:
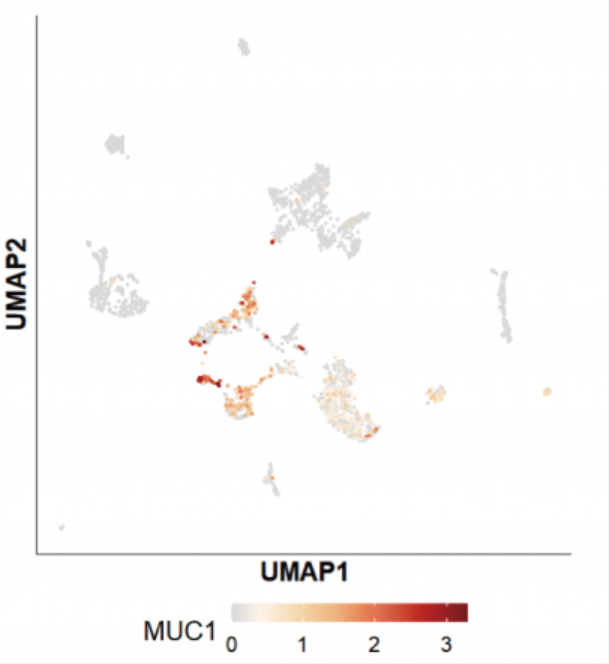
不过,该基因在三代全长测序中,并未测到该基因的转录本,下文有对此类现象的讨论。
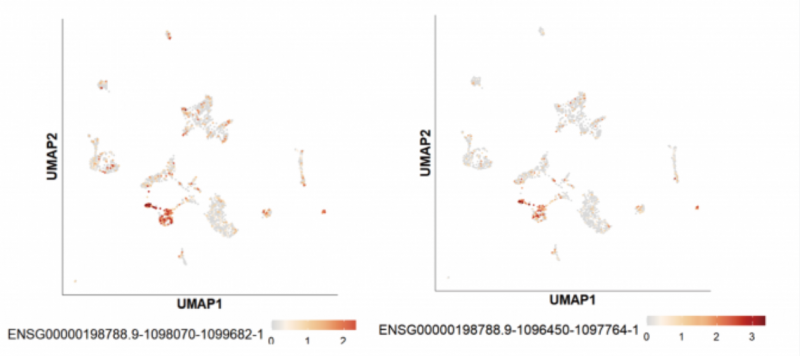
另外一个明星癌基因 MUC2 的表达展示:
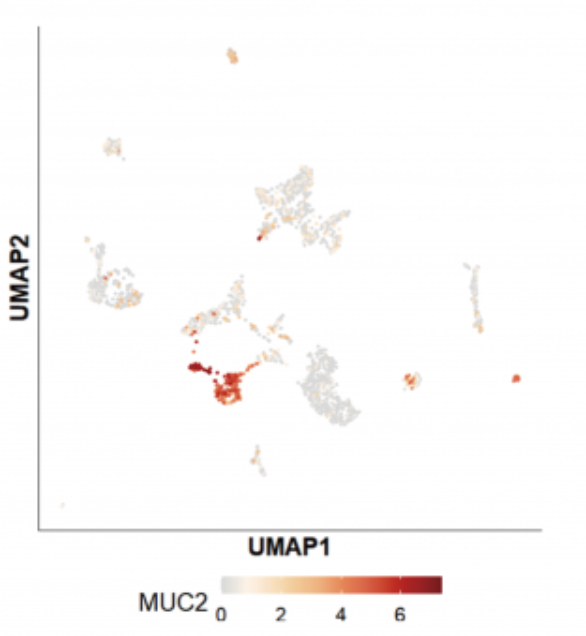
在三代全长数据中,MUC2 测到了多条 Ensemble 数据库中未收录的全新转录本:
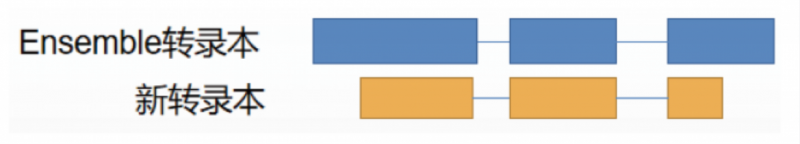
经过后续的序列比对(如下图红色区域),发现新转录本是已知转录本的一部分序列。

示例图如下:
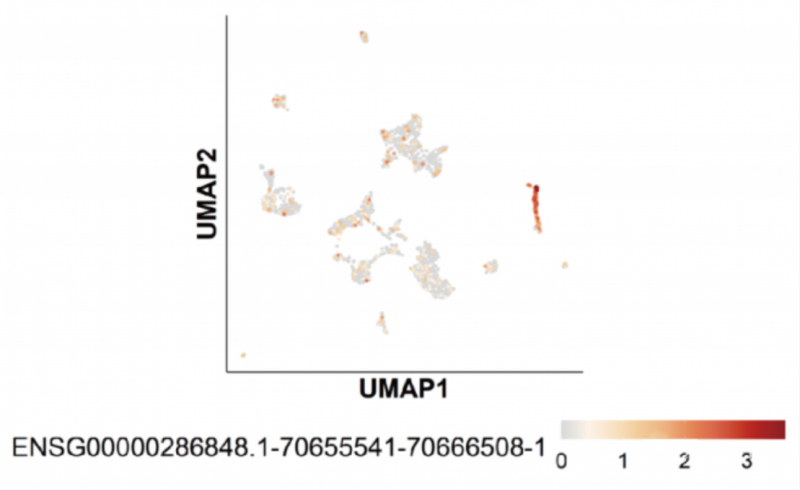
此外,全长转录本可以测到较多 lncRNA,下图为 B 细胞中某条表达特异性 lncRNA:
综合评价:
目前单细胞二代测序对基因进行表达定量的原理,是把该基因表达出来的所有转录本的 UM 进行求和,而三代测序是每条转录本进行单独计数,如某个基因在在细胞的 count 值是 100,理论上来说,在三代转录本数据中,可以对应找到 100 条转录本 UM1。不过,就实际情况来说,最终的数据产出都是依靠测序技术,现阶段的三代测序成本仍然较高,如果以比较高的投入来测非常多的三代数据是不太现实的。也就是说,只能在成本与高期望之间,选择一个比较适中的平衡。因此,是否选择使用单细胞全长测序技术,有如下几个建议:
1、传统的 Bulk-seq 研究中,借助 illumina、MGISEQ、Pacbio、Nanopore 等二代、三代测序技术同样可以测到较多的可变剪切、融合基因、新转录本等。唯一让研究者困扰的是测到的新序列太多(2000+),较难筛选过滤做后期功能验证。单细胞全长转录本测序的一个优势,是对新测到的转录本加上 细胞亚群 的标签,可以比较快速的筛选,如肿瘤细胞、免疫细胞亚群中的特异性新转录本,节省下游验证成本。这类课题是比较适合单细胞全长测序,最终结合实际数据发现特异性转录本。
2、如果已经指定了某个关键基因,想研究该基因是否在特定亚群中存在可变剪切或新转录本、融合基因等,比较建议先从已发表文章,或者公开数据库(如 htps:/singlecell.broadinstitute.ore/single_cell) 中,预先査询该基因的单细胞表达丰度。如果该基因表达量较低,并且亚群(如 T 细胞)的表达比例低于 50%,则要慎重选择单细胞全长测序方法。解决方法是可以把这群细胞通过流式、磁珠等技术分选出来(人为富集),单独对这群细胞进行单细胞二代 + 三代测序。
3、从上述的 MUC1 未检测到转录本,而 MUC2 检测到多条新转录本的例子来看,在现阶段较低测序投入情况下,三代全长技术更倾向于测出来高丰度的转录本。从另外一个角度来说,如果某条转录本出现在三代全长的表达矩阵中,也就意味着该转录本在真实细胞中的基础表达丰度仍然较高。这种方式相当于过滤掉了低丰度的转录本,这个对于 siRNA 干扰、膜蛋白、lncRNA 项目来说算是增加了个较为可信的属性。另外,如果一定要看 MUC1 基因的转录本,其实可以通过 IGV 导入二代测序的 BAM 文件,直接查看该基因的原始测序序列,可以在一定程度上通过已经测到的这些 reads 来探索该 reads 分属于哪条转录本(如果该基因存在多种转录本的话)。
4、单细胞三代全长可以直接给出来每条新转录本的序列,不过测序错误的情况也时常发生,所以后期验证必不可少。

1、高质量标准:严格按 ISO9001:2015 质量标准执行;
2、标准化内控:丰富的实操经验构建了标准化的内控体系;
3、流程化分析:完善的分析流程,准确快速解析单细胞转录本数据;
4、专业的团队:专业的技术团队具有多年项目方案设计、实验操作、售后分析等经验;
5、全流程服务:提供样本处理、建库测序及数据分析的全套服务。
伯豪生物提供:单细胞核测序、单细胞核测序技术服务

- 样本类型: 新鲜组织,原代细胞,细胞系等。
- 样本来源: 血液提取、磁珠富集、流式富集、组织解离等。
- 样本量及其它质控要求:
(1)细胞悬液:>10* 目标细胞个数(最少 10,000 个细胞);活率 >85%;浓度 500-1,000 个细胞 / ul;细胞间无粘连(成团率 <5%);无大于 40um 的细胞碎片或其它颗粒物;不存在逆转录抑制剂和非细胞的核酸分子。
(2)血液:EDTA 抗凝的全血(不可肝素抗凝),>5ml。
(3)组织:0.3 cm × 0.3 cm(不超过 0.5cm × 0.5cm)的新鲜组织,4~5 块。
- 样本保存运输:
(1)细胞悬液:最好现场制备,如要运输,建议使用细胞保护液,4°C 运输,48 小时内送达伯豪生物实验室。
(2)血液:EDTA 抗凝的全血,4°C 运输,4 小时内送达实验室;或提取 PBMC 后冻存,干冰运输。
(3)组织:建议使用单细胞专用的组织保护液,4°C 运输,48 小时内送达实验室。
- 捕获细胞数及测序数据量:
| 捕获细胞数 | 二代测序数据量 | 三代测序数据量 |
| 3000-6000(最佳建议) | 70-100G | 100G |
| 6000-8000 | 100G | 150G |
| 8000-11000 | 100G | 200G |
| 不建议超过 11000 细胞 |
- 全外测序 20G
伯豪生物提供:单细

| 序号 | 文件类型 | 查阅 |
| 1 | 【画册】单细胞全长转录本测序解决方案 | 点击查看 |
| 2 | 【画册】石蜡样本(FFPE)单细胞转录组测序解决方案 | 点击查看 |
| 3 | 【画册】单细胞测序解决方案 | 点击查看 |
