好不容易拿到了自己的空间转录组测序数据,心情激动、跃跃欲试,想赶快去实施一下自己的 idea,可是又不会写代码,只能干等着?当然不是,其实 空间转录组测序数据 你完全可以实现不写代码完成自己大部分数据挖掘的工作。
这里轮到有用的工具 10x genomic Loupe Browser 上场了!Loupe Browser 是 10x genomics 专门开发的用于 10x 相关产品可视化的工具,它其实可以完成很大部分的数据挖掘工作,而且操作简单,自己有 空间转录组测序数据 或单细胞测序数据的确实可以好好利用一下这个软件。
Loupe Browser 下载地址:
https://support.10xgenomics.com/spatial-gene-expression/software/downloads/latest?
下载后直接双击安装就可。
数据准备
Loupe Browser 导入的是 Space Ranger 软件生成的 cloupe.cloupe 文件。10x genomic Space Ranger 软件的使用教程可参考:
空间转录组第二讲:Space Ranger 的使用
前面也介绍过,我们做空间转录组测序一般不太可能只做一个样本,一般会 做空间转录组测序 多个样本同时分析,因为需要进行 空间转录组测序目标 亚群数目和基因表达差异的比较,这时候就需要把 空间转录组测序数据 多个样本整合起来一起分析。用 Loupe Browser 挖掘 数据也是一样,尽量使用多样本整合后的 cloupe.cloupe 文件。
10x genomic 的软件 spacerangeraggr 来合并多个空间转录组测序的样本使用教程可参考:
空间转录组第五讲:10x spaceranger aggr 合并多个样本
如果前面用 seurat 对数据进行来了分析,我们也可以把 seurat 聚类的结果导入到 Loupe Browser 中进行数据后续的挖掘(cloupe.cloupe 文件也是需要准备的)。
软件操作介绍
一、文件读取和 Loupe Browser 整体界面介绍
文件读取可以选择简单的方式,就是双击自己的 cloupe.cloupe 文件,当然前提是你已经安装好 Loupe Browser 软件。然后会进入如下界面:
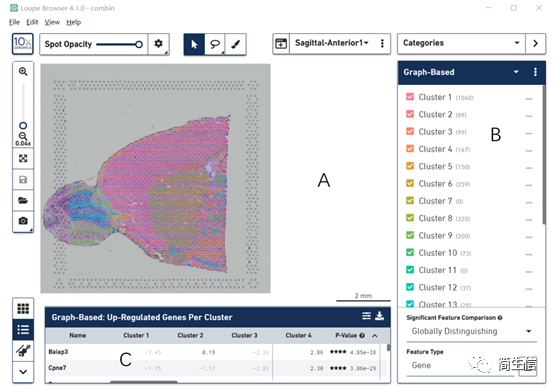
A 区为图像展示和操作区,可以对组织图像进行操作,也可以展示降维聚类后的 tsne 或 umap 分布图。
B 区主要对亚群、基因、样本选择进行操作。
C 区展示差异基因的结果。
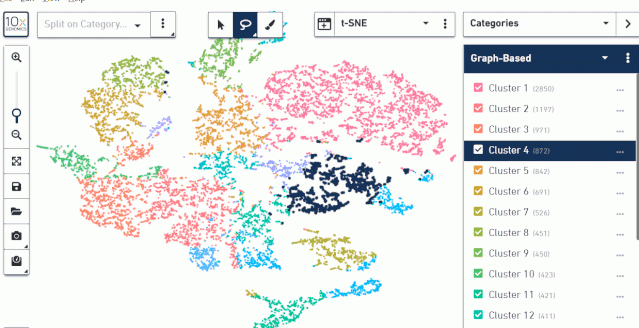
二、查看亚群总体分布
这里可以查看 tsne(umap)图亚群的分布,也可以查看每个样本组织切片上亚群的分布。
三、对图像区域进行操作
主要包括 3 个工具,箭头展示对应点的亚群信息,套索可以选中某个区域进行操作,可以进行分类或导出,后一个画笔可以对单个点进行选择,也可以进行分类或导出。
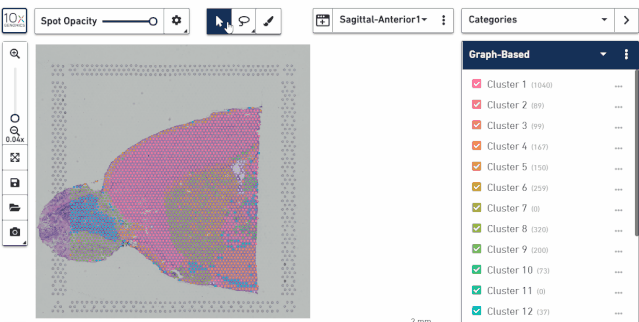
在这里我们用套索工具挑选了两群细胞,同时又用画笔给第二群细胞增加了几个细胞,因为有时候我们想把个别分散的点加到亚群里去,这个用套索工具是没办法实现的。
根据组织切片染色来圈范围。有时候我们只需要跟组织染色的结果来进行分组划分区域,这时候显示 spot 点的信息反而会使组织图像看不清楚,这里可以使用 spot opacity 工具来调整 spot 的透明度,甚至完全去掉亚群信息,然后再根据组织切片图像进行区域的选择。
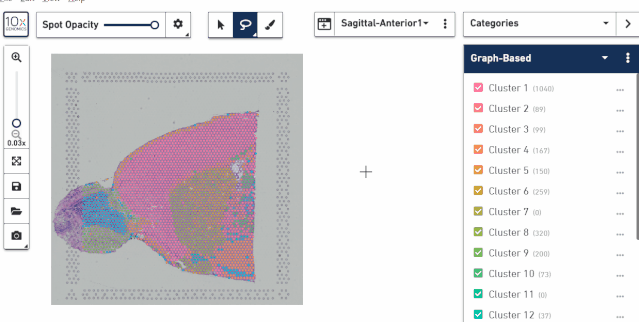
四、对亚群进行选择
在右边区域对亚群进行选择,可以选中自己想要查看的亚群,然后展示该亚群在样本组织图和 tsne(或 umap)降维聚类图中的分布。
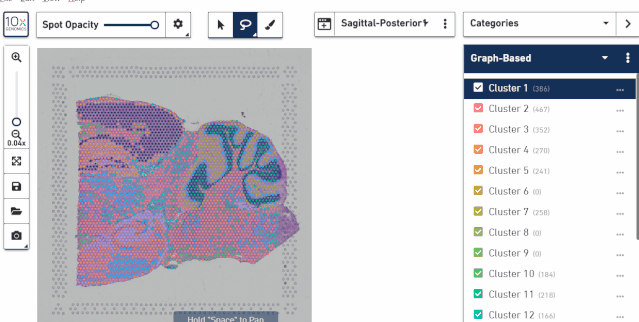
五、对样本进行选择
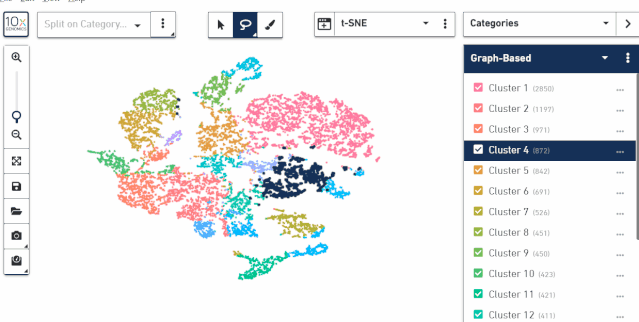
对于数据 tsne 或 umap 的可视化结果同样可以对样本进行选择展示,查看每个样本的位置分布。
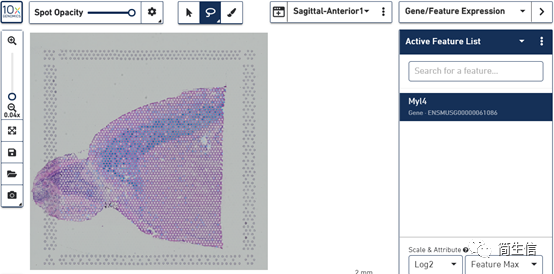
六、查看基因表达
右边上方选择 Gene/Feature Eexression,然后下面空白框中输入需要查看的基因,就可以查看这个基因在亚群以及样本中的表达分布情况。
七、Marker 基因查看
在界面的下方,可以查看每个亚群对应的 marker 基因信息包括 P 值、FC 等,也可以把 marker 基因差异结果表格导出来。
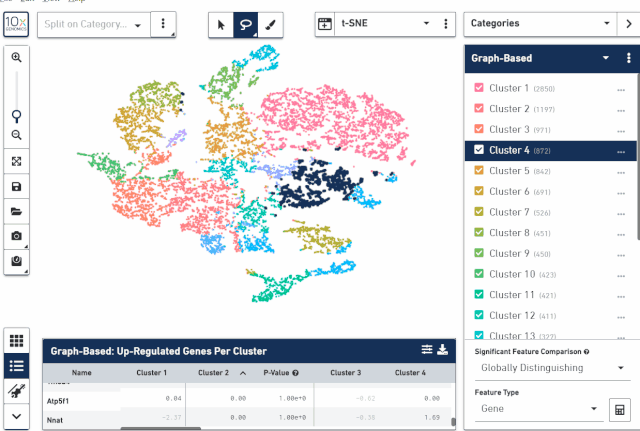
从上面的操作中可以看发现,不但可以查看和导出亚群上调的基因,也可以导出下调的基因,基因数目则可以选择 top20、50、100 或者所有基因。
导出的 marker 基因表格如下:
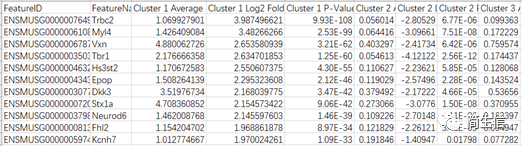
虽然我们只选择某个亚群的 marker 基因,但是实际上软件会把这些基因在所有亚群中的差异信息值都导出来。主要包括三列:亚群的平均表达值、log2 foldchage、pvalue。
八、导入自己的 tsne 降维和分类结果
前面我们看到的亚群分类和 tsne 降维结果都是 10x spaceranger 软件自己计算出来的,有时候我们自己用其他软件(比如 Seurat)分析后得到的结果也想用 Loupe Browser 软件来进行可视化和数据的挖掘,这时候可以选择将自己的亚群分类结果和 tsne 降维坐标信息导入软件内,替代原有的亚群分类和 tsne 降维展示。
文件准备
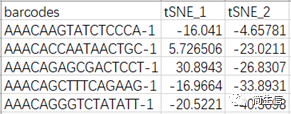
tsne 坐标文件 data_tsne.csv:包括 3 列信息(barcode、tSNE_1、tSNE_2)
Cluster 分群文件 data_cluster.csv,除了分群信息我们也可以加入样本分组信息。
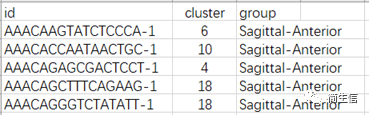
注意文件必须是 csv 格式,且 barcode 的 id 要与 10x spaceranger 跑出的结果一致。
导入文件
开始数据挖掘
重点来咯,敲黑板啦!
前面介绍了 Loupe Browser 的基本操作,下面来介绍一下怎么利用该软件进行有效的空间转录组测序数据挖掘。
一、亚群聚类结果的选择
从前面的介绍我们知道这里既可以使用 10xspaceranger 软件聚类的结果,也可以导入 Seurat 聚类的结果。如果同时有着两种结果可以选择,那么我们可以挑选一个更优的结果进行后面的分析。理论上这两种聚类的结果我们都可以选择用来进行后续的分析,那么哪个更好就需要自己来判断一下那种结果更符合自己的预期了。可以从几个方面来判断:
A、样本的分布情况:一般来说如果聚类后如果样本之间没有交集互相独立,这样的结果不是很理想的,说明没有有效的去除个体差异。但是因为空转的数据比较特殊,本身不同切片不同区域很难做到 RNA 捕获的均一性,有时候不同样本的数据差异就是很大,强行通过归一化或其他方法去除个体差异反而会使结果失真。
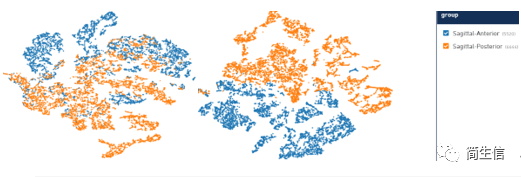
注:这个示例图左边是 spaceranger 的结果,右边是 Seurat 聚类的结果,单从样本分布来看 spaceranger 的结果是更佳的。
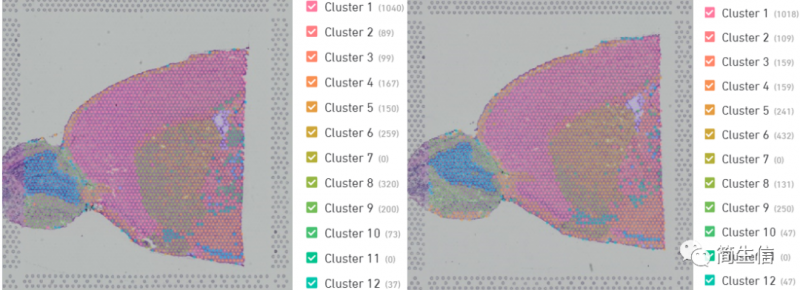
B、结合组织切片染色结果来判断:组织切片区域的构成、病理状态的分布对于判断亚群的分布是否符合预期可能更有用。比如说从组织切片上已经知道某个区域就是属于某一类细胞,那么这一区域的细胞聚成一类的结果肯定更合适的。

注:示例图里 spaceranger 的结果(左边)相对来说比 seurat 的聚类结果(中间)更符合组织切片上的纹路。
二、亚群分布比较
我们拿到数据的首步,一般会先看一下不同亚群在不同样本里的分布情况,哪些亚群是共有的,哪些亚群是样本特有的,哪些亚群数目变化比较大的。如果有做生物学重复还可以看一下重复性效果怎么样。由于软件只能一个样本一个样本的查看,这时候我们可以把图片截图下来放到一起来展示。对于亚群数目的比较,如果自己可以写代码用图形化展示出来肯定是先进的,如果不会写代码也可以把亚群对应的数字输入到 excle 表格里直接进行统计。

从示例图例我们可以发现两种切片的生物学重复还是很好的。7、11 号群是 posterior 样本特有的,6、8、9 号群是 anterior 样本特有的。后面我们也可以重点关注这些群到底属于什么细胞类型。
三、细胞类型注释
空间转录组测序技术不是真正的单细胞水平,每个 spot 会捕获 5 -10 个细胞,这样每个 spot 里实际上可能存在几种不同类型的细胞。但是对于大部分组织细胞来说同一区域周围更可能分布着相同类型的细胞,这样对应的 spot 孔里面更容易捕获到同一种细胞(或者 splot 里的大部分细胞属于同一类型)。所以对空间转录组测序数据进行细胞类型注释有利判断不同组织细胞类型的大致空间分布信息。对于免疫细胞要研究它的空间分布往往是比较困难的,它常常会散布整个组织上,而且聚类的时候也比较难得到集中的免疫细胞群。
做亚群细胞类型注释的方法一般有两种,一种是用专门的软件去做注释(如 singleR),还有一种就是根据已知 maker 基因的表达来对亚群进行注释判断。这里我们采用第二种方法。
细胞类型 marker 基因来源
细胞类型 marker 基因的可以是自己从文献中收集的,也可是从一些单细胞 marker 基因数据库里找来的。这里我们主要来介绍怎么使用 CellMarker 数据库里的细胞 marker 基因来做注释。CellMarker 数据库收录了 158 种组织 / 亚组织的 467 种人细胞类型,81 种组织 / 亚组织的 389 种鼠细胞类型。数据主要来源于文献和数据库,包括单细胞测序数据和生物实验数据。
网址:http://biocc.hrbmu.edu.cn/CellMarker/
数据库主界面:
我们的示例数据是小鼠的,这里我们点击小鼠图标,出现下面界面。
选择组织类型:这里我们选小鼠脑,鼠标点击脑的图标会出来对应的细胞类型,一共 22 种细胞。
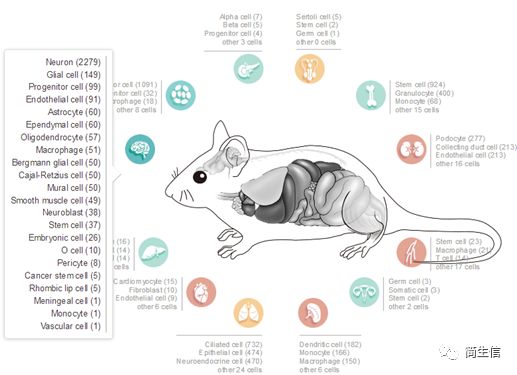
然后点击某个细胞类型会进入该细胞类型 marker 基因的界面,例如点击星形胶质细胞,出现 Astrocyte 细胞的 marker 基因词云图。

字体越大表示标志物生物学证据越多,右边有标志物生物学证据数目的排序。一般我们选择 3 - 5 个排名靠前的 marker 基因来注释细胞就好了,太多反而容易造成干扰。这里我们选择前 3 个基因 Gfap、Aldh1l1、Atp1b2 来进行星形胶质细胞的注释。
Loupe Browser 展示 marker 基因
按照前面的操作说明,右边上方选择 Gene/Feature Eexression,然后下面空白框中输入需要查看的基因。为了方便查看基因在每个亚群里的表达可以使用 Loupe Browser 的网格分割的展示方式。
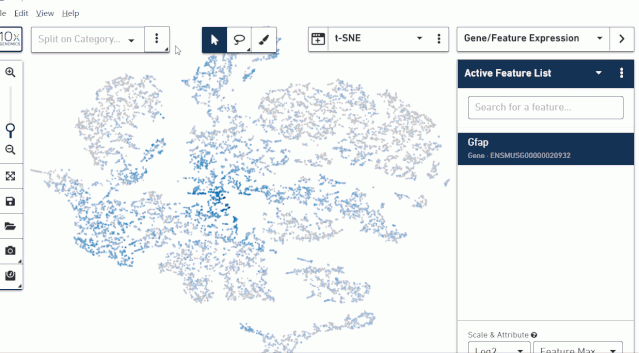
我们可以把几个基因的结果截图下来合并到一起来分析。有时候蓝色看起来表达差异不明显,也可以点击软件右下角的颜色工具替换色系。
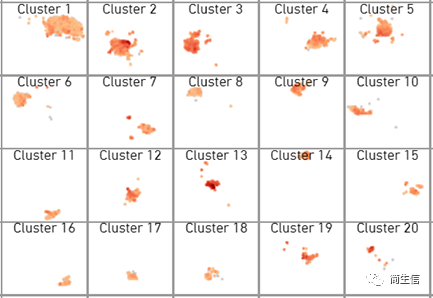
GFAP
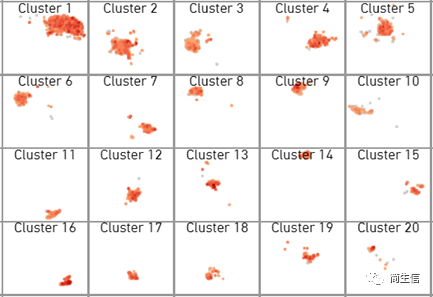
Aldh1l1
Atp1b2
看到这 3 个基因的表达分布图,基因之间的表达分布并不是那么一致,尤其是第 3 个基因都看不出哪个亚群高哪个亚群低。这种情况在空转数据或单细胞数据中是经常会出现的。这时候我们一般优先参考排序靠前的也就是更经典的 marker 基因的结果。GFAP 是星形胶质细胞经典的 marker,从它表达分布来看 2 号和 13 群的表达更高一点,尤其是 13 号群。从第二个 marker 基因 Aldh1l1 的表达来看 13 号群也相对更高一点。所以我们先暂定这 13 号群为 Astrocyte 细胞群。
细胞类型辅助判断方法
有时候用上面的方法我们还不能完全确认某个亚群的细胞类型,这时候我们可以借助第二种方法进一步判断,就是利用自己数据亚群的 marker 基因来分析。这里我们首先把 13 号群的 marker 基因表格导出来,前面已经讲述了导出亚群 marker 基因的方法。
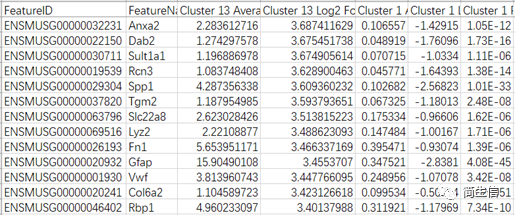
步骤一:看亚群 marker 基因的交集
这里我们发现 GFAP 确实是 13 号亚群的特异的 marker 基因,且平均表达值和 log2FC 还挺大的。
步骤二:看亚群 marker 基因富集到的功能
这里利用 KOBAS 3.0 进行富集分析,这个软件使用起来很简单,几乎看一眼就会,而且它 3.0 版本 2019 年进行了更新,里面收录的数据库也比较全比较新。
网站:http://kobas.cbi.pku.edu.cn/kobas3/genelist/
选择物种(这里选择小鼠),把 marker 基因 gene symbol 复制粘贴进去。
选择用来富集的库,这里我们选择 GO 功能可能更有利于细胞类型的判断。
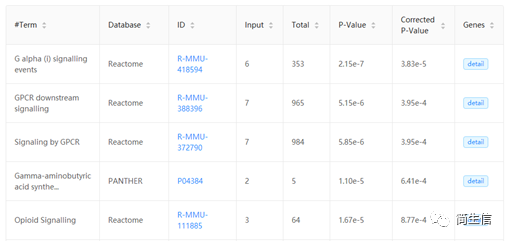
点击 run 提交,等待几分钟出现下面界面:
点击 download 下载结果文件,结果表格如下:
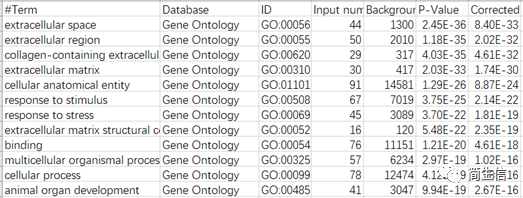
我们通过文献或资料先找到 Astrocyte 细胞细胞的主要功能,然后再看富集结果中是否正好富集到这些功能,这样可以帮助我们进一步确认亚群的注释结果是否正确。
修改亚群名称
确认好亚群的细胞类型之后,我们就可以在 Loupe Browser 上直接修改 lable 了。
该类型的细胞分布展示
当我们确定了亚群属于什么细胞之后,接着可以来查看这一细胞类型在组织图片上的分布了。从这上面也许我们也能找到一些有价值的信息。
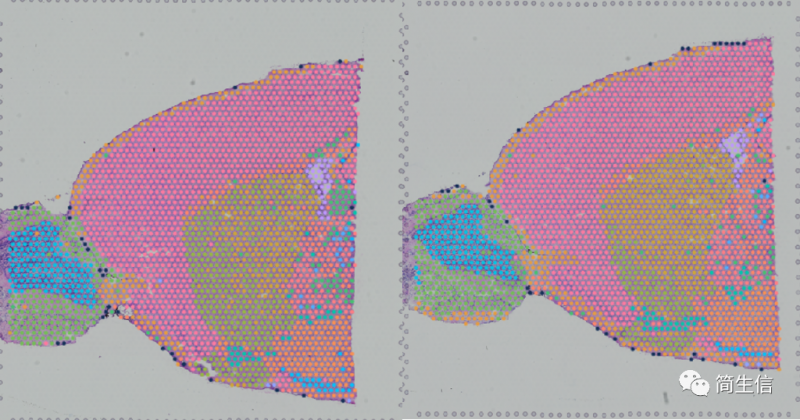
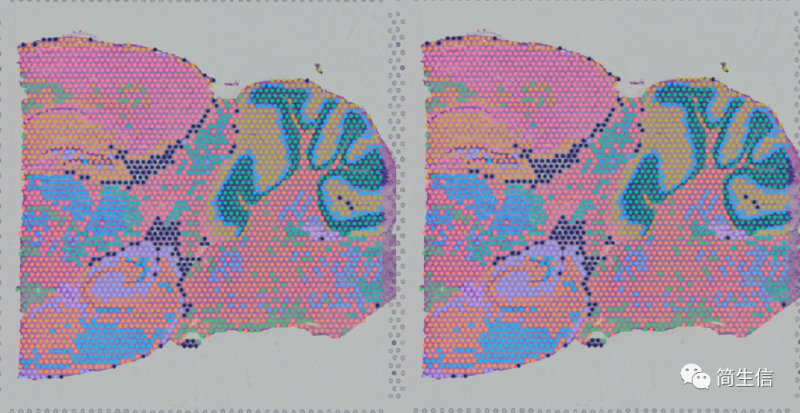
这里我们发现星形胶质细胞的分布其实还蛮有意思的。
四、细胞亚群在不同分组中的差异分析
当我们找到自己关注的细胞类型或亚群之后,下一步就可以去分析这种细胞类型(或亚群)在不同组织处理或是不同病例状态下的基因差异和功能差异。可以是样本之间比较,也可以是样本分组之后的比较。比如说比较肿瘤原发灶和转移灶上皮细胞基因表达的差异。这里我们就用分析前矢状面(Sagittal-Anterior)和后矢状面(Sagittal-Posterior)亚群的差异来展示一下方法。
进行差异分析之前我们需要先手动制作分类文件,因为 LoupeBrowser 需要根据选择的分组来进行差异分析。先导出亚群分类表格,接着导出细胞样本对应表格,再将两个表格进行合并来设置分类。后将做好的分类文件重新导入 Loupe Browser 中。
制作分组文件表格
这里我们比较 13 号亚群 Astrocyte 细胞作为示例来展示怎么分析差异。新做好的分组文件如下,把 13 号群的细胞分成了 Sagittal-Anterior_Astrocyte 和 Sagittal-Posterior_Astrocyte 两组。注意文件存为 csv 格式。

分析差异
接着把分组文件导入到 Loupe Browser 中,利用 Loupe Browser 右下角的计算机工具计算两组的差异,分析 Astrocyte 细胞在两组之间的差异基因。
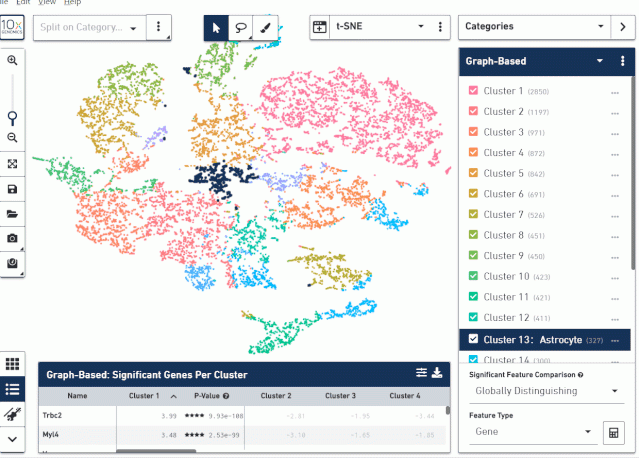
注意:因为我们只是想分析这两组之间的差异,所以右下角的 SignificantFeature Comparison 选择 Locally Distinguishing,否则会计算出来这两个分组相对于所有细胞之间的差异基因。
功能富集
后我们可以将前面得到的差异基因用 KOBAS 3.0 进行富集分析,分析 Astrocyte 细胞在两组之间的功能差异。
五、结合组织区域分布对数据进行挖掘
大部分组织其实是有其特定的区域划分的,比如说大脑里有皮层、丘脑、海马、脉络丛等多个区域。将组织的区域划分和亚群(或细胞类型)的分布结合起来还是能发现很多有价值的信息的。
组织分区
可以根据这些区域特异表达的 maker 基因的分布来判断每个区域在组织切片上的位置。
皮层 marker 基因 STX1A 的表达分布:
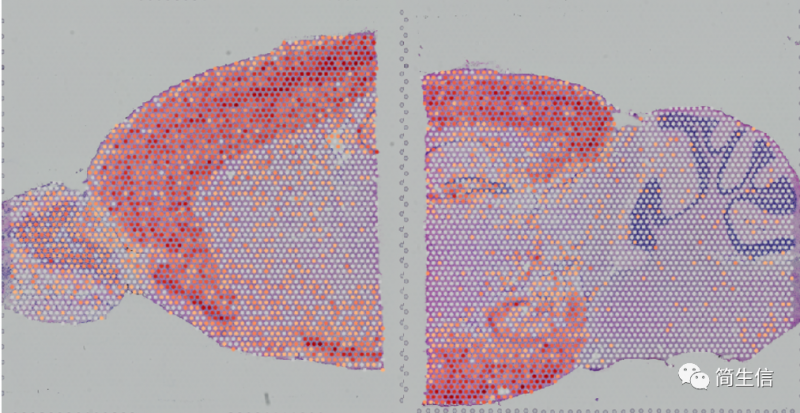
丘脑 marker 基因 PRKCD 的表达分布:
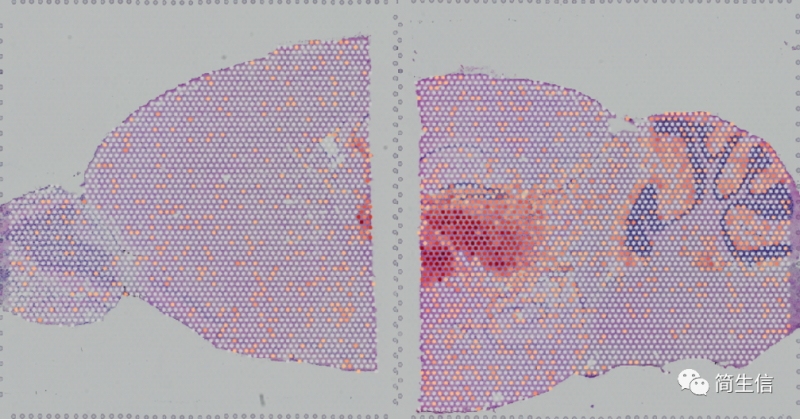
海马 marker 基因 HPCA 的表达分布:
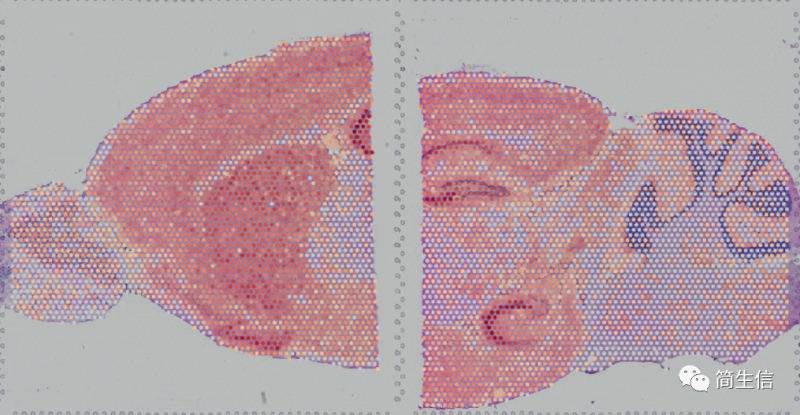
脉络丛 marker 基因 TTR 的表达分布:
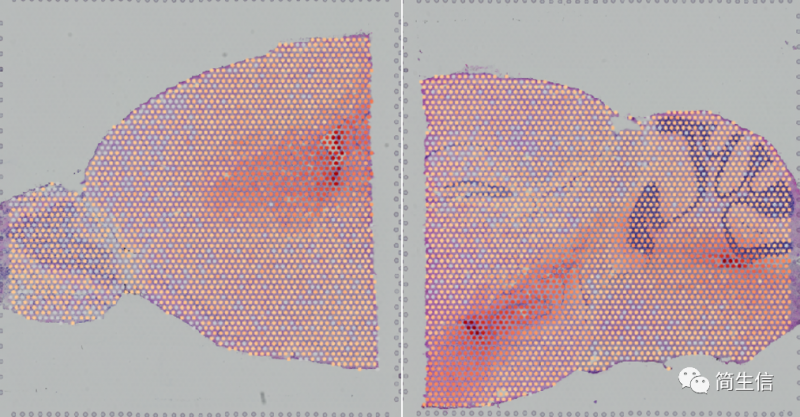
不同区域的亚群分布
找到了对应的区域之后,下一步就可以研究每个区域主要有哪些亚群,包括哪些细胞类型,不同区域之间细胞类型之间的差异,不同区域之间功能的差异。
将这个数据的区域分布图和亚群分布图结合起来看的时候其实能发现一些挺有意思的现象。1、4、15 号群基本上都分布在皮层,17 号群对应丘脑,脉络丛对应 20 号群。
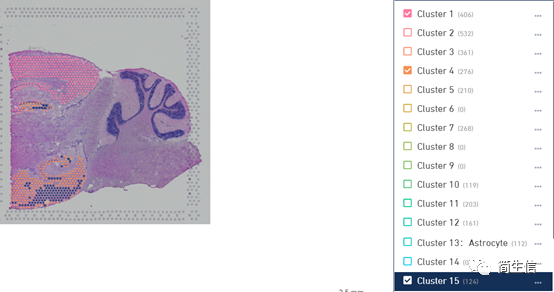
选中 1、4、15 号群

选中 17 号群

选中 20 号群
这里如果有多个样本分组的话(病理状态、疾病分期等等),则可以统计在不同分组下每二个区域亚群的分布情况,比如说皮层区在正常状态下 1 号亚群起主要作用,在疾病状态下 4 号亚群起主要作用。
Marker 基因和功能研究
除了前面说的分析亚群的分布情况,我们还可以分析亚群或整个区域的功能变化。比如说分析脉络丛对应的 20 亚群在正常状态和疾病状态下特异表达的 maker 基因以及功能的变化。具体的操作方法跟前面分组差异分析相同,这里不再演示操作步骤。
六、结合病理学特征对数据进行挖掘
空间转录组测序技术正真的精髓不是研究细胞亚群的分布,而在于将它在空间位置上体现的异质性跟组织病理学特征的分布进行结合,挖掘在不同病理学特征下转录组学的差异。这对于研究疾病病变的机制、帮助临床实现更好的患者分子分型、以及空间位置 Biomarker 的挖掘方面都是非常有价值的。
比如说我们的组织切片上同时分布着不同严重程度(或不同类型)的病灶区,我们可以手动把这些区域圈出来进行转录组层面的比较,找出不同病灶区的特异性 marker,分析疾病在一步步发展进程中生物学功能的变化,甚至可以思考一下是否能找出一些关键性因子来阻断疾病的进展。
当然也可以结合前面细胞类型注释结果,分析组织切片不同病理学特征下某一类细胞的功能学的差异,前提条件是这类细胞在组织上的分布是比较集中的,可以清晰的从图像上找出来的。
因为我们的示例数据没有病例学信息,这里我们随意选择两个区域进行具体操作演示。首先利用上面中间的套索工具选择不同病理学特征对应的区域进行命名分组,然后利用坐下角计算器工具计算每组特异性的 marker 基因。
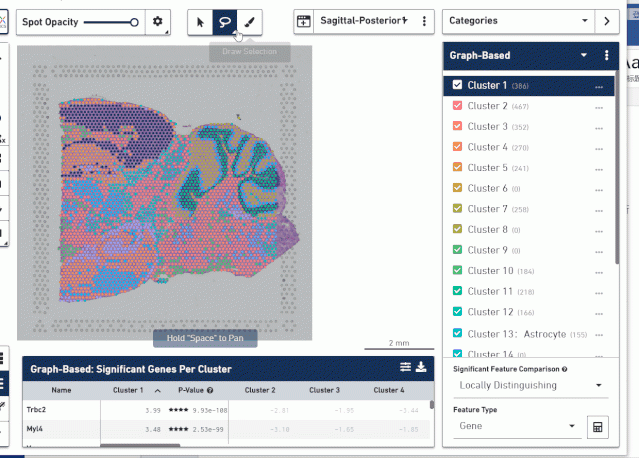
导出差异基因接着用 KOBAS 3.0 进行富集分析,分析这些基因主要富集到哪些功能上。
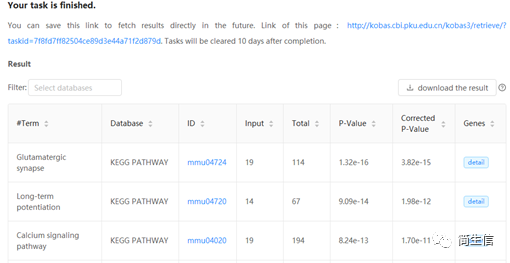
得到了差异基因和功能富集的表格,接下来就需要自己认真的去里面挖掘有价值的信息了。
伯豪生物提供从样本采集至生信全范围覆盖的空间转录组测序技术服务,2020 年空间转录组测序技术刚刚兴起之时,空间转录组测序技术逐渐进入科研工作者视野,特别是临床肿瘤、细胞免疫等领域的应用,未来伯豪生物将继续为广大科研概及临床工作者提供高效优质的空间转录组测序技术服务。
更多伯豪生物人工服务:

