
Fungal–Bacterial Networks in the Habitat of SongRong (Tricholoma matsutake) and Driving Factors of Their Distribution Rules

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site Selection
2.2. Sampling Strategy and Processing
2.3. DNA Extraction and Illumina NovaSeq Platform
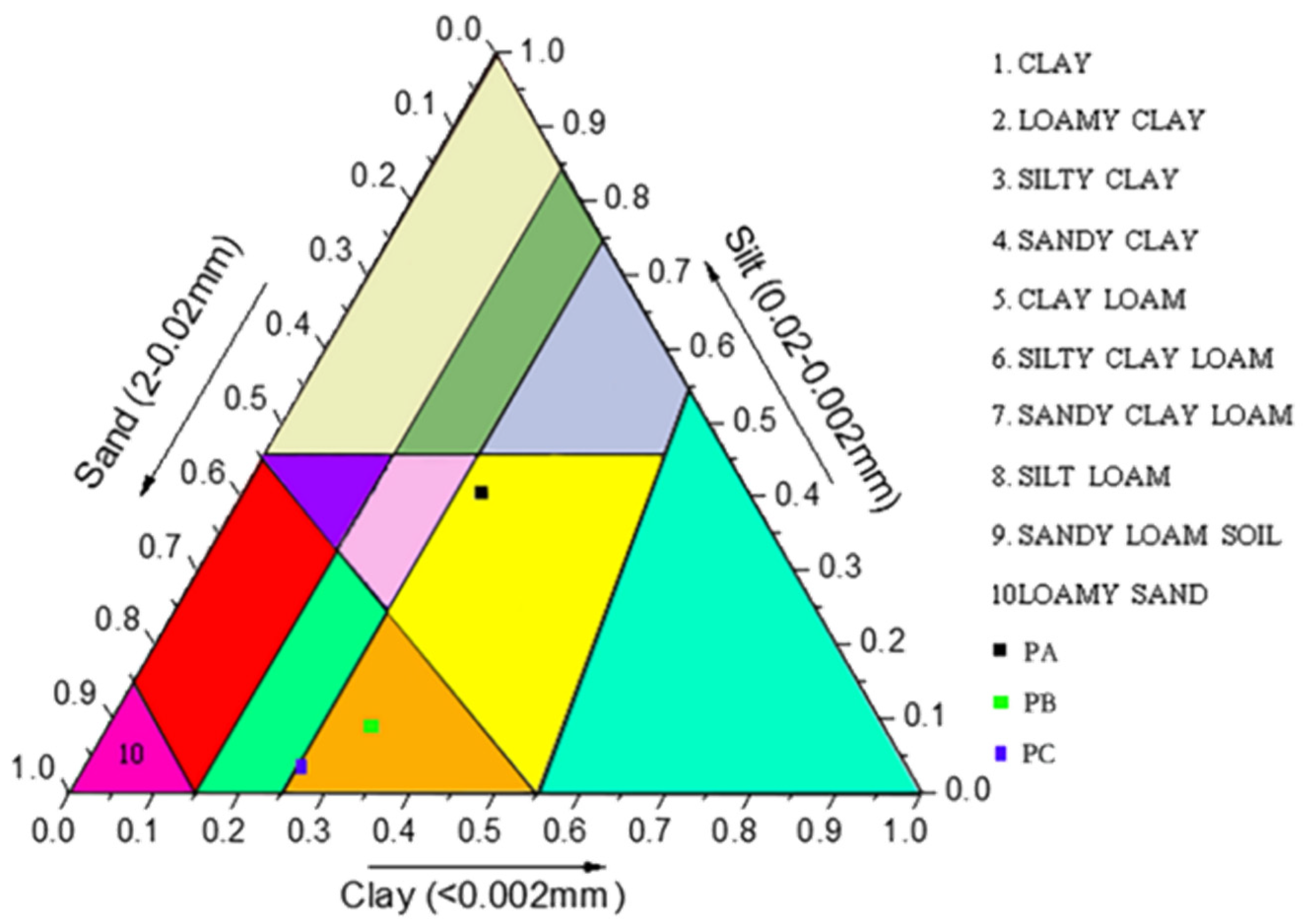
2.4. Analysis of Physical/Chemical Parameters of Soil Samples
2.5. Bioinformatics
2.6. Data Processing and Statistical Analysis
2.7. Fungal and Bacterial Communities
3. Results
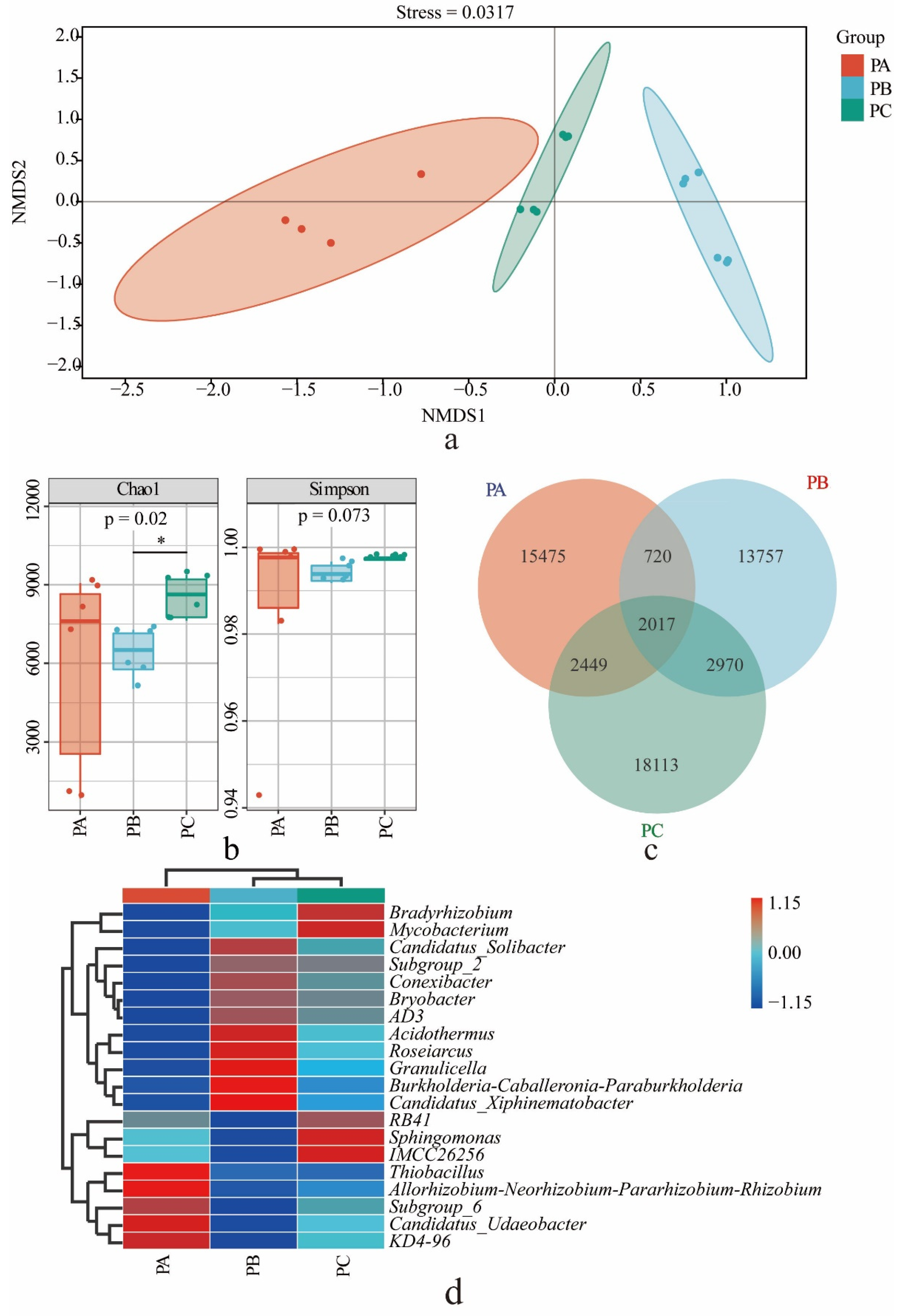
3.1. Bacterial Community in the Soil of T. matsutake Habitat
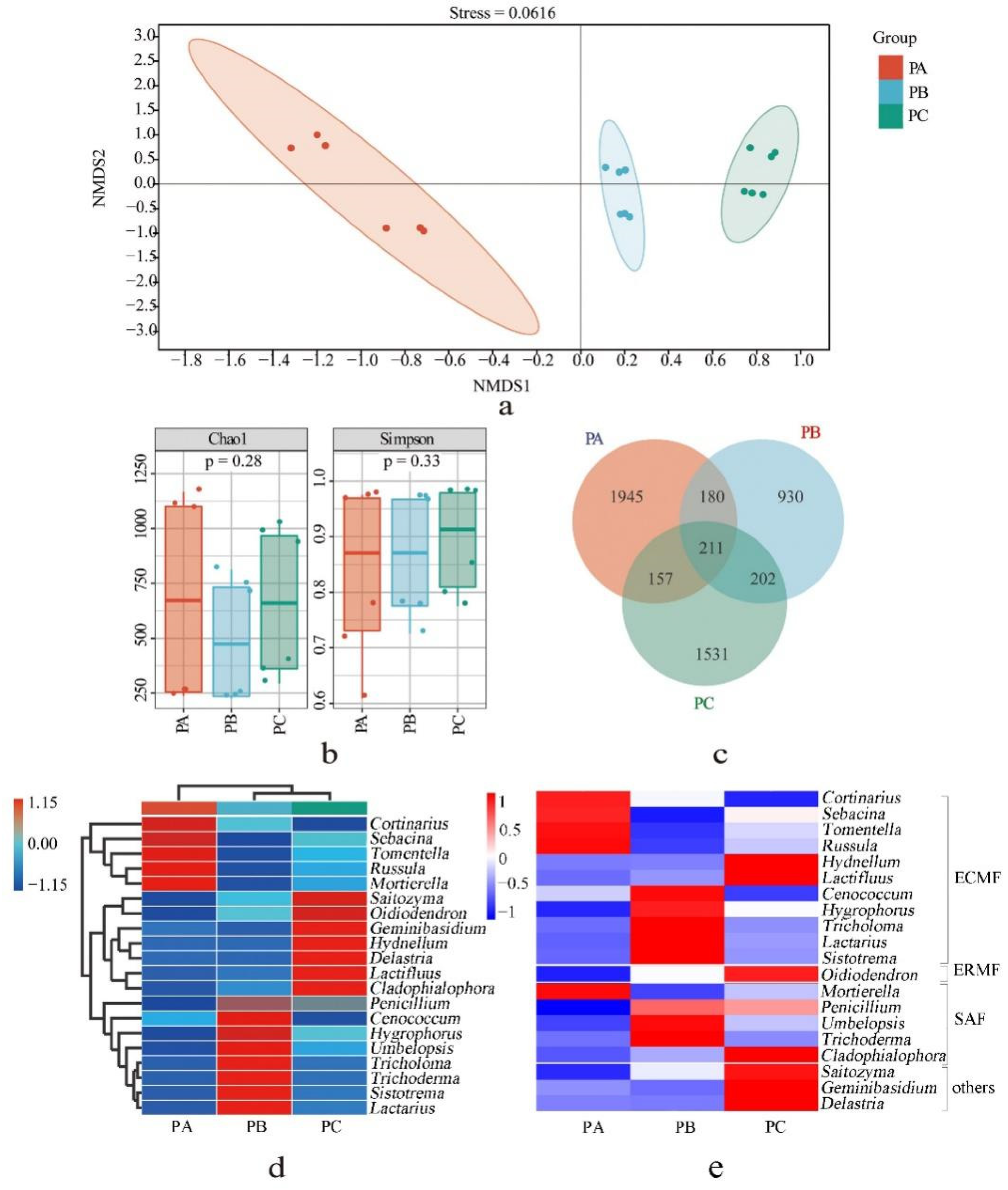
3.2. Fungal Community in the Soil of T. matsutake Habitat
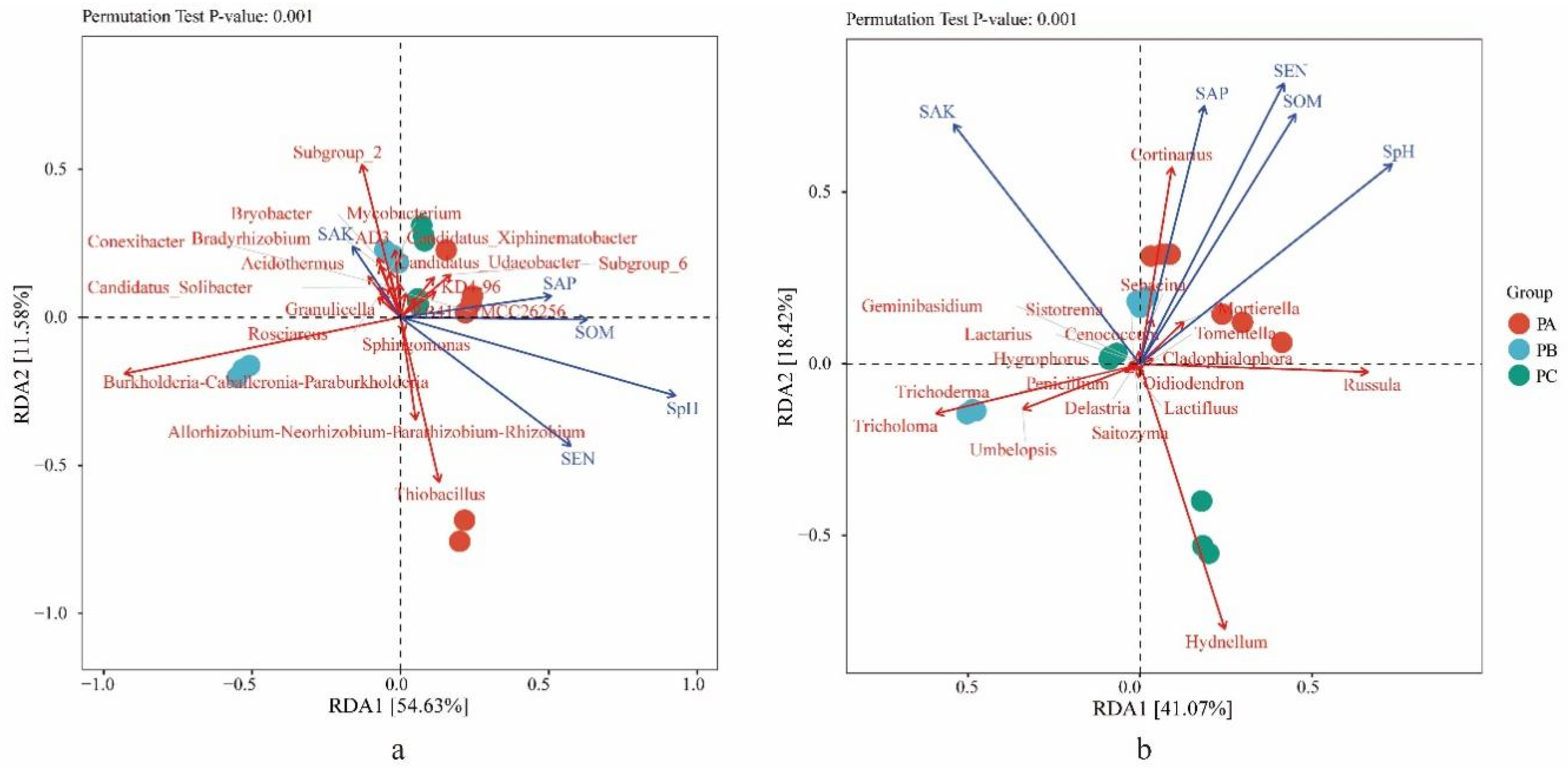
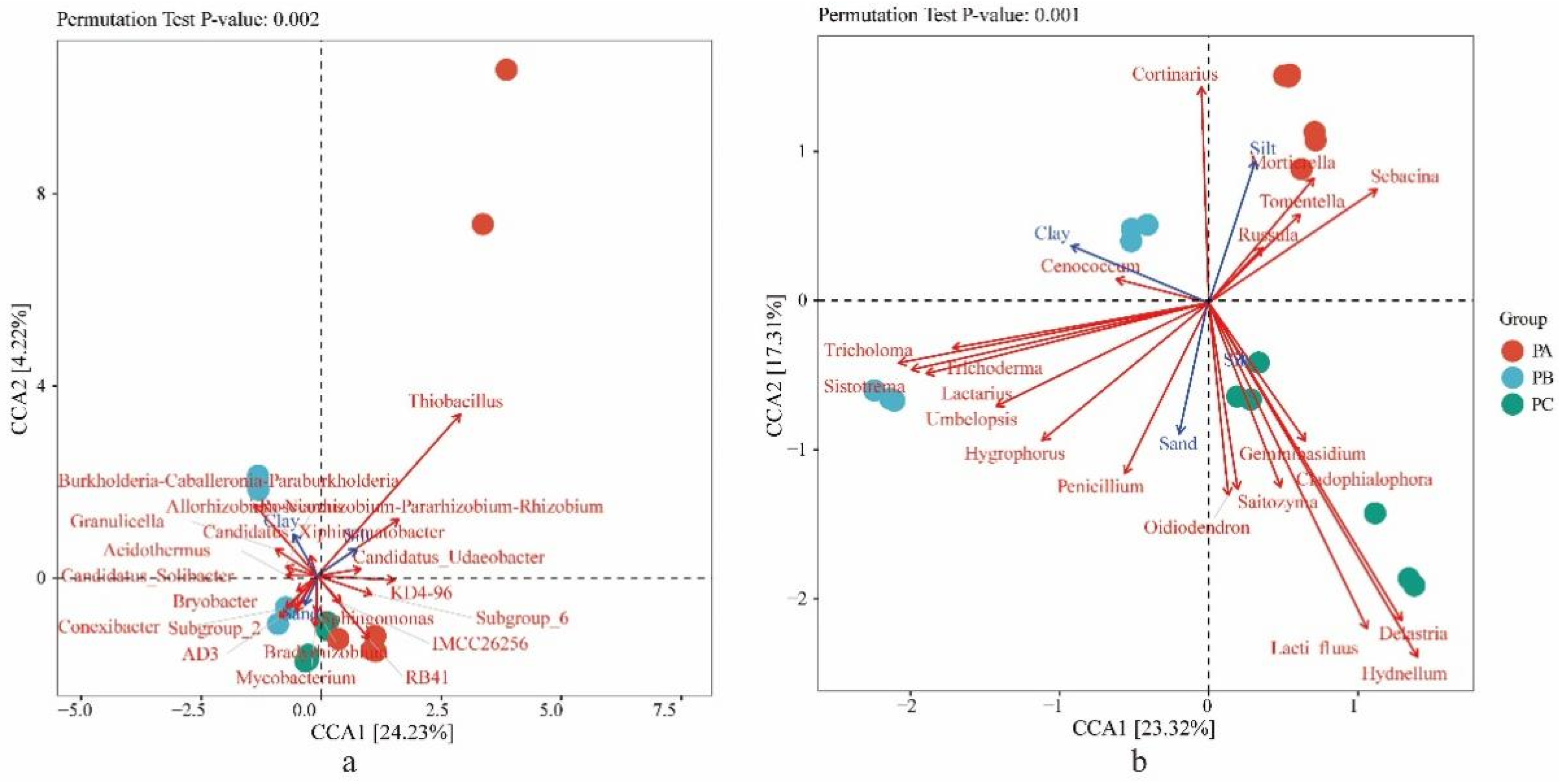
3.3. Soil Physicochemical Conditions and Their Effects on the Distributions of the Fungal and Bacterial Community
3.4. The Community of Tricholoma in Three Collection Sites and the Influence Factors
3.5. Functional Potential Analysis of Soil Fungi and Bacteria in the Habitat of T. matsutake
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Yu, F.Q.; Zhang, C.X.; Li, S.H. Tricholoma matsutake: An edible mycorrhizal mushroom of high socioeconomic relevance in China. Rev. Mex. Micol. 2017, 46, 55–61. [Google Scholar]
- Amend, A.; Garbelotto, M.; Fang, Z.D.; Keeley, S. Isolation by landscape in populations of a prized edible mushroom Tricholoma matsutake. Conserv. Genet. 2010, 11, 795–802. [Google Scholar] [CrossRef]
- Yao, Y.J.; Wei, J.C.; Zhuang, W.Y.; Cai, L.; Liu, D.M.; Li, J.S.; Li, Y.; Wang, K.; Wu, H.J. Development of red list assessment of macrofungi in China. Sheng Wu Duo Yang Xing 2020, 28, 4. [Google Scholar] [CrossRef]
- Xian, M.Y. Distribution and ecological environment of Tricholoma matsutake in Sichuan Province. Edible Fungi 1989, 5, 9–10. [Google Scholar]
- Liao, S.Y.; Leng, H.Q.; Liu, B. The special ecological environment of matsutake mushroom in Sichuan. J. Sichuan Agric. Univ. 1991, 2, 297–302. [Google Scholar]
- Gong, M.Q.; Chen, Y. A study on Pinus and Eucalyptus ectomycorrhiza in south China. For. Res. 1991, 3, 323–327. [Google Scholar]
- Amend, A.; Keeley, S.; Garbelotto, M. Forest age correlates with fine-scale spatial structure of matsutake mycorrhizas. Mycol. Res. 2009, 113, 541–551. [Google Scholar] [CrossRef]
- Takashi, Y.; Akiyoshi, Y.; Hitoshi, F. Advances in the cultivation of the highly-prized ectomycorrhizal mushroom Tricholoma matsutake. Mycoscience 2020, 61, 49–57. [Google Scholar] [CrossRef]
- Li, Q.; Li, X.L.; Huang, W.L.; Xiong, C.; Yang, Y.; Yang, Z.R.; Zheng, L.Y. Community structure and diversity of entophytic bacteria in Tricholoma matsutake in Sichuan Province, Southwest China. Ying Yong Sheng Tai Xue Bao 2014, 25, 3316–3322. [Google Scholar] [CrossRef]
- Voříšková, J.; Brabcová, V.; Cajthaml, T.; Baldrian, P. Seasonal dynamics of fungal communities in a temperate oak forest soil. New Phytol. 2014, 201, 269–278. [Google Scholar] [CrossRef]
- Xing, P.J.; Xu, Y.; Gao, T.T.; Li, G.L.; Zhou, J.J.; Xie, M.L.; Ji, R.Q. The community composition variation of Russulaceae associated with the Quercus mongolica forest during the growing season at Wudalianchi City, China. PeerJ 2020, 8, e8527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.T.; Li, G.L.; Zhou, J.J.; Ji, R.Q.; Meng, L.P. Fungi community structure associated with Korean pine forests and the varying tendency with four forest land age. Nova Hedwig. 2020, 111, 391–415. [Google Scholar] [CrossRef]
- Roy, M.; Schimann, H.; Braga-Neto, R.; Da Silva, R.A.E.; Duque, J.; Frame, D.; Wartchow, F.; Neves, M.A. Diversity and distribution of ectomycorrhizal fungi from Amazonian lowland white-sand forests in Brazil and French Guiana. Biotropica 2016, 48, 90–100. [Google Scholar] [CrossRef]
- Park, M.S.; Oh, S.Y.; Cho, H.J.; Fong, J.J.; Cheon, W.J.; Lim, Y.W. Trichoderma songyi sp. nov., a new species associated with the pine mushroom (Tricholoma matsutake). Antonie Van Leeuwenhoek 2014, 106, 593–603. [Google Scholar] [CrossRef]
- Saravesi, K.; Markkola, A.; Taulavuori, E.; Syvänperä, I.; Suominen, O.; Suokas, M.; Saikkonend, K.; Taulavuoria, K. Impacts of experimental warming and northern light climate on growth and root fungal communities of Scots pine populations. Fungal Ecol. 2019, 40, 43–49. [Google Scholar] [CrossRef]
- Fu, W.J.; Xu, G.B.; Liu, J.S.; Lian, Y.J. Research on distribution and ecological environment of Tricholoma matsutake in Changbai Mountain area. Acta Edulis Fungi 1996, 3, 48. Available online: http://www.syjxb.com/EN/Y1996/V03/I03/48 (accessed on 29 April 2022).
- Tan, W. Cultivation theory and method of Trichotoma matsutake. Acta Edulis Fungi 1994, 1, 53–63. [Google Scholar] [CrossRef]
- Matsushita, N.; Shindo, K.; Kikuchi, K.; Vaario, L.-M.; Suzuki, K. Ribosomal DNA diversity of Tricholoma matstuake in Yunnan, China. In Proceedings of the 115th Annual Japanese Forestry Society Meeting, Tokyo, Japan, 31 March–4 April 2004; Volume 4, p. 656. (In Japanese). [Google Scholar]
- Matsushita, N.; Kikuchi, K.; Sasaki, Y.; Guerin-Laguette, A.; Vaario, L.M.; Suzuki, K.; Intini, M. Genetic relationship of Tricholoma matsutake and T. nauseosum from the Northern Hemisphere based on analyses of ribosomal DNA spacer regions. Mycoscience 2005, 46, 90–96. [Google Scholar] [CrossRef]
- Masui, K. A study of the ectotrophic mycorrhizas of woody plants. Mem. Coll. Sci. Kyoto Imp. Univ. Ser. B 1927, 3, 149–279. [Google Scholar] [CrossRef]
- Murata, H.; Babasaki, K.; Yamada, A. Highly polymorphic DNA markers to specify strains of the ectomycorrhizal basidiomycete Tricholoma matsutake based on σmarY1, the long terminal repeat of gypsy-type retroelement marY1. Mycorrhiza 2005, 15, 179–186. [Google Scholar] [CrossRef]
- Yamada, A.; Endo, N.; Murata, H.; Ohta, A.; Fukuda, M. Tricholoma matsutake Y1 strain associated with Pinus densiflora shows a gradient of in vitro ectomycorrhizal specificity with Pinaceae and oak hosts. Mycoscience 2014, 55, 27–34. [Google Scholar] [CrossRef]
- Vaario, L.M.; Pennanen, T.; Lu, J.R.; Palmén, J.; Stenman, J.; Leveinen, J.; Kilpeläinen, P.; Kitunen, V. Tricholoma matsutake can absorb and accumulate trace elements directly from rock fragments in the shiro. Mycorrhiza 2015, 25, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Yamada, A.; Maruyama, T.; Neda, H. Ectomycorrhizas in vitro between Tricholoma matsutake, a basidiomycete that associates with Pinaceae, and Betula platyphylla var. japonica, an early successional birch species, in cool-temperate forests. Mycorrhiza 2015, 25, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Lim, Y.W. Effect of fairy ring bacteria on the growth of Tricholoma matsutake in vitro culture. Mycorrhiza 2018, 28, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.Q.; Xie, M.L.; Li, G.L.; Xu, Y.; Gao, T.T.; Xing, P.J.; Meng, L.P.; Liu, S.Y. Response of bacterial community structure to different ecological niches and their functions in Korean pine forests. PeerJ 2022, 10, e12978. [Google Scholar] [CrossRef] [PubMed]
- Lladó, S.; Lopez-Mondejar, R.; Baldrian, P. Drivers of microbial community structure in forest soils. Appl. Microbiol. Biotechnol. 2018, 102, 4331–4338. [Google Scholar] [CrossRef]
- Ogawa, M. Microbial ecology of mycorrhizal fungus, Tricholoma matsutake Ito et Imai (Sing.) in pine forest. I. Fungal colony (Shiro) of Tricholoma matsutake. Bull. Gov. For. Exp. Stn. 1975, 272, 79–121. [Google Scholar]
- Fu, W.J.; Xu, G.B.; Fu, M.J.; Liang, Y.J.; Liu, W.L.; Quang, X.L. Advance of Tricholoma matsutake in the area of Changbai Mountain and formation of the Shiro theory. Sci. Found. China 2006, 1, 8–10. [Google Scholar] [CrossRef]
- Laura, M.S.; Nadia, B.; Sue, B.; Hans, P.D.; Karl, D.F.; Richards, F.; García, P.; Gehrman, J.; Kristöfel, F.; Manninger, M.; et al. Environmental drivers of ectomycorrhizal communities in Europe’s temperate oak forests. Mol. Ecol. 2014, 23, 5628–5644. [Google Scholar] [CrossRef]
- Köhler, J.; Yang, N.; Pena, R.; Raghavan, V.; Polle, A.; Meier, I.C. Ectomycorrhizal fungal diversity increases phosphorus uptake efficiency of European beech. New Phytol. 2018, 220, 1200–1210. [Google Scholar] [CrossRef]
- Hassink, J.; Bouwman, L.A.; Zwart, K.B.; Bloem, J.; Brussaard, L. Relationships between soil texture, physical protection of organic matter, soil biota, and C and N mineralization in grassland soils. Geoderma 1993, 57, 105–128. [Google Scholar] [CrossRef]
- Hassink, J.; Bouwman, L.A.; Zwart, K.B.; Brussaard, L. Relationships between habitable pore space, soil biota and mineralization rates in grassland soils. Soil Biol. Biochem. 1993, 25, 47–55. [Google Scholar] [CrossRef]
- Ritz, K.; Young, I.M. Interactions between soil structure and fungi. Mycologist 2004, 18, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Erktan, A.; Or, D.; Scheu, S. The physical structure of soil: Determinant and consequence of trophic interactions. Soil Biol. Biochem. 2020, 148, 107876. [Google Scholar] [CrossRef]
- Guo, J.J.; Liu, W.B.; Zhu, C.; Luo, G.W.; Kong, Y.L.; Ling, N.; Wang, M.; Dai, J.Y.; Shen, Q.R.; Guo, S.W. Bacterial rather than fungal community composition is associated with microbial activities and nutrient-use efficiencies in a paddy soil with short-term organic amendments. Plant Soil 2018, 424, 335–349. [Google Scholar] [CrossRef]
- Yang, M.H.; Yang, X.M.; Chen, L.G. Studies on the relationship between Tricholoma matsutake and other rhizosphere microorganisms. Acta Agric. Univ. Jiangxiensis 1997, 4, 78–82. [Google Scholar]
- Ohara, H.; Hamada, M. Disappearance of bacteria from the zone of active mycorrhizas in Tricholoma matsutake (S. Ito et Imai) Singer. Nature 1967, 213, 528–529. [Google Scholar] [CrossRef]
- Li, Q.; Li, X.L.; Chen, C.; Li, S.H.; Huang, W.L.; Xiong, C.; Jin, X.; Zheng, L.Y. Analysis of bacterial diversity and communities associated with Tricholoma matsutake fruiting bodies by barcoded pyrosequencing in Sichuan province, southwest China. J. Microbiol. Biotechnol. 2016, 26, 89–98. [Google Scholar] [CrossRef]
- Garbaye, J. Mycorrhiza helper bacteria: A new dimension to the mycorrhizal symbiosis. New Phytol. 1994, 128, 197–210. [Google Scholar] [CrossRef]
- Yu, X.; Yu, K.; Chen, B.; Liao, Z.H.; Liang, J.Y.; Yao, Q.C.; Qin, Z.J.; Wang, H.; Yu, J. Different responses of scleractinian coral Acropora pruinosa from Weizhou Island during extreme high temperature events. Coral Reefs 2021, 40, 1697–1711. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Bao, S.D. Soil Agrochemical Analysis, 3rd ed.; China Agricultural Press: Beijing, China, 2000; pp. 1–495. [Google Scholar]
- Zhou, J.M.; Shen, R.F. Dictionary of Soil Science; Science Press: Beijing, China, 2013; pp. 1–902. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Prepr. 2018, 6, e27295v1. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Hamilton, N. An extension to ‘ggplot2’, for the creation of ternary diagrams. Anesthesiology 2015, 64, 72–86. [Google Scholar]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, W.; Wei, J. Ectomycorrhizal fungal community in the rhizospheric soil of Betula platyphylla in Inner Mongolia. Mycosystema 2018, 37, 294–304. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2: An improved and extensible approach for metagenome inference. bioRxiv 2019, 672295, preprint. [Google Scholar] [CrossRef] [Green Version]
- Suz, L.M.; Kallow, S.; Reed, K.; Bidartondo, M.I.; Barsoum, N. Pine mycorrhizal communities in pure and mixed pine-oak forests: Abiotic environment trumps neighboring oak host effects. For. Ecol. Manag. 2017, 406, 370–380. [Google Scholar] [CrossRef]
- Xiong, C.; Zhu, Y.G.; Wang, J.T.; Singh, B.; Han, L.L.; Shen, J.P.; Li, P.P.; Wang, G.B.; Wu, C.F.; Ge, A.H.; et al. Host selection shapes crop microbiome assembly and network complexity. New Phytol. 2021, 229, 1091–1104. [Google Scholar] [CrossRef]
- Yamanaka, T.; Ota, Y.; Konno, M.; Kawai, M.; Ohta, A.; Neda, H.; Terashima, Y.; Yamada, A. The host ranges of cutonifer-associated Tricholoma matsutake, Fagaceae-associated T. bakamatsutake and T. fulvocastaneum are wider in vitro than in nature. Mycology 2014, 106, 397–406. [Google Scholar] [CrossRef]
- Zechmeister-Boltenstern, S.; Michel, K.; Pfeffer, M. Soil microbial community structure in European forests in relation to forest type and atmospheric nitrogen deposition. Plant Soil 2011, 343, 37–50. [Google Scholar] [CrossRef]
- Genre, A.; Lanfranco, L.; Perotto, S.; Bonfante, P. Unique and common traits in mycorrhizal symbioses. Nat. Rev. Microbiol. 2020, 18, 649–660. [Google Scholar] [CrossRef]
- Fang, M.; Li, Y.; Yao, F.J.; Ni, S.F. The Summarize of Research on Tricholoma matsutake. Chin. Edible Fungi 2005, 6, 12–15. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeanbille, M.; Buée, M.; Bach, C.; Cébron, A.; Frey-Klett, P.; Turpault, M.P.; Uroz, S. Soil parameters drive the structure, diversity and metabolic potentials of the bacterial communities across temperate beech forest soil sequences. Microb. Ecol. 2016, 71, 482–493. [Google Scholar] [CrossRef]
- Bonito, G.; Benucci, G.M.N.; Hameed, K.; Weighill, D.; Jones, P.; Chen, K.H.; Jacobson, D.; Schadt, C.; Vilgalys, R. Fungal-Bacterial networks in the Populus rhizobiome are impacted by soil properties and host genotype. Front. Microbiol. 2019, 10, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hereira-Pacheco, S.E.; Navarro-Noya, Y.E.; Dendooven, L. The root endophytic bacterial community of Ricinus communis L. resembles the seeds community more than the rhizosphere bacteria independent of soil water content. Sci. Rep. 2021, 11, e2173. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Oh, S.Y.; Yoo, S.; Fong, J.J.; Kim, C.S.; Jo, J.W.; Lim, Y.W. Influence of Season and Soil Properties on Fungal Communities of Neighboring Climax Forests (Carpinus cordata and Fraxinus rhynchophylla). Front. Microbiol. 2020, 11, 572706. [Google Scholar] [CrossRef]
- Wang, Y.L.; Gao, C.; Chen, L.; Ji, N.N.; Wu, B.W.; Li, X.C.; Lü, P.P.; Zheng, Y.; Guo, L.D. Host plant phylogeny and geographic distance strongly structure betulaceae-associated ectomycorrhizal fungal communities in Chinese secondary forest ecosystems. FEMS Microbiol. Ecol. 2019, 4, fiz037. [Google Scholar] [CrossRef]
- Wang, X.B.; Lv, X.T.; Yao, J.; Wang, Z.W.; Deng, Y.; Cheng, W.X.; Zhou, J.Z.; Han, X.G. Habitat-specific patterns and drivers of bacterial β-diversity in China’s drylands. ISME J. 2017, 11, 1345–1358. [Google Scholar] [CrossRef]
- Bahram, M.; Hildebrand, F.; Forslund, S.K.; Anderson, J.L.; Soudzilovskaia, N.A.; Bodegom, P.M.; Bengtsson-Palme, J.; Anslan, S.; Coelho, L.P.; Harend, H.; et al. Structure and function of the global topsoil microbiome. Nature 2018, 560, 233–237. [Google Scholar] [CrossRef]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microb. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [Green Version]
- Demoling, F.; Nilsson, L.O.; Bååth, E. Bacterial and fungal response to nitrogen fertilization in three coniferous forest soils. Soil Biol. Biochem. 2008, 40, 370–379. [Google Scholar] [CrossRef]
- Xiong, W.; Jousset, A.; Guo, S.; Karlsson, I.; Zhao, Q.Y.; Wu, H.S.; Kowalchuk, G.A.; Shen, Q.R.; Li, R.; Geisen, S. Soil protist communities form a dynamic hub in the soil microbiome. ISME J. 2018, 12, 634–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurgel, S.N.; Douglas, G.M.; Dusault, A.; Percival, D.; Langille, M.G.I. Dissecting community structure in wild blueberry root and soil microbiome. Front. Microbiol. 2018, 9, 1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, R.; Siddiqui, Z.A.; Kikuchi, J.; Ando, M.; Sriwati, R.; Nozaki, A.; Futai, K. Detecting nonculturable bacteria in the active mycorrhizal zone of the pine mushroom Tricholoma matsutake. J. Microbiol. 2012, 50, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Sutarman, S.; Jalaluddin, A.K.; Li’aini, A.S.; Prihatiningrum, A.E. Characterizations of Trichoderma sp. and its effect on Ralstonia solanacearum of tobacco seedlings. J. HPT Trop. 2021, 21, 8–19. [Google Scholar] [CrossRef]
- Jeong, M.; Choi, D.H.; Cheon, W.J.; Kim, J.G. Pyrosequencing and taxonomic composition of the fungal community from soil of Tricholoma matsutake in Gyeongju. J. Microbiol. Biotechnol. 2021, 31, 686–695. [Google Scholar] [CrossRef]
- Zhou, J.; Gui, H.; Yang, S.; Yang, X.; Shi, L. Fungal interactions matter: Tricholoma matsutake domination affect fungal diversity and function in mountain forest soils. Biology 2021, 10, 1051. [Google Scholar] [CrossRef]
- Fujii, S.; Unezaki, S.; Okumura, T.; Miura, R.; Saito, K. Asparagine-linked carbohydrate of penicillium notatum phospholipase b. J. Biochem. 1994, 116, 204–208. [Google Scholar] [CrossRef]
- Labbé, J.L.; Weston, D.J.; Dunkirk, N.; Pelletier, D.A.; Tuskan, G.A. Newly identified helper bacteria stimulate ectomycorrhizal formation in Populus. Front. Plant Sci. 2014, 5, 579. [Google Scholar] [CrossRef]
- Delgado, M.J.; Casella, S.; Bedmar, E.J. Denitrification in rhizobia-legume symbiosis. In Biology of the Nitrogen Cycle; Bothe, H., Ferguson, S.J., Newton, W.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 83–91. [Google Scholar] [CrossRef]
- Alvarez-Santullano, N.; Villegas, P.; Mardones, M.S.; Durán, R.E.; Donoso, R.; González, A.; Sanhueza, C.; Navia, R.; Acevedo, F.; Pérez-Pantoja, D.; et al. Genome-wide metabolic reconstruction of the synthesis of Polyhydroxyalkanoates from sugars and fatty acids by Burkholderia sensu lato species. Microorganisms 2021, 9, 1290. [Google Scholar] [CrossRef]
- Johnsen, U.; Sutter, J.M.; Zaiß, H.; Schönheit, P. L-Arabinose degradation pathway in the haloarchaeon Haloferax volcanii involves a novel type of l-arabinose dehydrogenase. Extremophiles 2013, 17, 897–909. [Google Scholar] [CrossRef]
- Pedrosa, F.O.; Zancan, G.T. L-Arabinose metabolism in Rhizobium japonicum. J. Bacteriol. 1974, 119, 336–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Shimada, N.; Tajima, K.; Kodaki, T.; Makino, K. Identification and characterization of L-arabonate dehydratase, L-2-keto-3-deoxyarabonate dehydratase, and L-arabinolactonase involved in an alternative pathway of L-arabinose metabolism. Novel evolutionary insight into sugar metabolism. J. Biol. Chem. 2006, 281, 33521–33536. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SpH | SOM (mg·kg−1) | SAK (mg·kg−1) | SEN (mg·kg−1) | SAP (mg·kg−1) | |
|---|---|---|---|---|---|
| PA | 5.71 ± 0.07 a | 106,893.17 ± 9456.2 a | 23.57 ± 3.37 a | 203.73 ± 9.26 a | 20.20 ± 6.34 a |
| PB | 5.00 ± 0.12 c | 68,044.84 ± 5118.48 b | 25.83 ± 0.58 a | 114.32 ± 4.48 b | 5.19 ± 1.31 b |
| PC | 5.31 ± 0.06 b | 56,300.90 ± 5525.63 b | 20.27 ± 1.85 a | 94.82 ± 3.06 c | 3.87 ± 1.01 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, R.-Q.; Xu, Y.; Si, Y.-J.; Phukhamsakda, C.; Li, Y.; Meng, L.-P.; Liu, S.-Y.; Xie, M.-L. Fungal–Bacterial Networks in the Habitat of SongRong (Tricholoma matsutake) and Driving Factors of Their Distribution Rules. J. Fungi 2022, 8, 575. https://doi.org/10.3390/jof8060575
Ji R-Q, Xu Y, Si Y-J, Phukhamsakda C, Li Y, Meng L-P, Liu S-Y, Xie M-L. Fungal–Bacterial Networks in the Habitat of SongRong (Tricholoma matsutake) and Driving Factors of Their Distribution Rules. Journal of Fungi. 2022; 8(6):575. https://doi.org/10.3390/jof8060575
Chicago/Turabian StyleJi, Rui-Qing, Yang Xu, Yan-Ji Si, Chayanard Phukhamsakda, Yu Li, Li-Peng Meng, Shu-Yan Liu, and Meng-Le Xie. 2022. "Fungal–Bacterial Networks in the Habitat of SongRong (Tricholoma matsutake) and Driving Factors of Their Distribution Rules" Journal of Fungi 8, no. 6: 575. https://doi.org/10.3390/jof8060575