Q: 目前常用的预测 miR 的靶基因的工具网站有哪些?
A:miRWalk、TargetScan、DIANA-microT、MicroCosm、miRDB、miRanda、PITA。其中 TargetScan 及 PITA 不支持大鼠。
A:miRWalk、TargetScan、DIANA-microT、MicroCosm、miRDB、miRanda、PITA。其中 TargetScan 及 PITA 不支持大鼠。
A:【查看】
拟南芥 【查看】
A:1ug,纯化后 3ug。
A:不建议,如果起始量足够可以尝试。
A:全基因组覆盖,得到更多 CpG 位点信息,还能获得 CHG、CHH 位点的甲基化信息。

A:只有有参考基因组的物种都可以做 WGBS,非常适合于基因组较小的植物。
A:通常建议测基因组大小 *30 的数据量,比如人的参考基因大小为 3G, 建议测序 90G.
A:可以,伯豪积累了许多相关经验,还可以将甲基化与 mRNA、lncRNA 分别进行关联分析。可参考附件。【查看】
A:1ng,纯化后 30ng。
A:片段过长可能与蛋白本身结合的 DNA 片段就偏大有关,就伯豪已做的 ChIP-seq 项目经验来看,片段过长对后续结果分析是不会有影响的。后续建库会对大片段 DNA 进行再次打断至 300 bp。
A:input 对照,Input 对照不仅可以验证染色质断裂的效果,还可以根据 Input 中的靶序列的含量以及染色质沉淀中的靶序列的含量,按照取样比例换算出 ChIP 的效率。
阳性对照,一般用 anti-RNA Polymerase II 抗体(阳性抗体),RNA Polymerase II 是通用转录因子,在所有细胞中都能结合基因(为了保证阳性对照有效,一般选择阳性对照的引物是根据管家基因设计的)的核心启动子区,因此,理论上 ChIP 后 PCR 都会有条带。
阴性对照,用普通的 IgG 做为阴性抗体,理论上不会 ChIP 下来任何 DNA 片段,但是由于非特异结合,或者实验过程中没发生结合的 DNA 清除不完全,可能也会出现比较浅的条带。
一个完整的 ChIP-seq 数据通常应该包含 input 对照或阴性对照,一般推荐好的有 input 对照。
A:【查看】
A:【查看】
A:【查看】
A:【查看】
A:基于超高重 PCR 的扩增目标区域后重测序,可以针对连续区段、外显子靶标以及客户指定区域,特异性设计扩增引物,经过引物优化及修饰,多重 PCR 一次性捕获所有位点。只要有参考序列即可,没有物种限制。
Q:目标区域重测序技术参数?
A: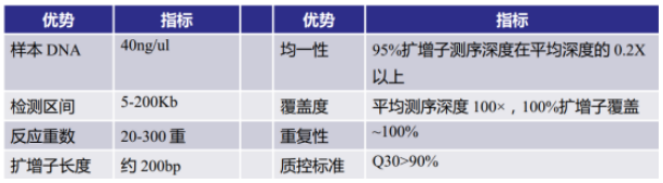
Q:目标区域重测序的样本要求?
A:基因组 DNA,要求:DNA 浓度≥40ng/ul,体积 >20ul。纯度 OD260/OD280 在 1.8~2.0 之间。
A:500ng。DNA 浓度不低于 55ng/μl,OD 260/280 值应在 1.7~2.0 之间,A260/A230 > 1.5;RNA 应该去除干净;不含有其它个体或其它物种的 DNA 污染。
A:DNA Qubit 定量满足大于 80ng,弥散带在 500bp 左右,并且无明显污染即可。如提供 FFPE,需提供切片厚度为 10μm 左右的 FFPE 切片 10 片左右。如保存年限太久、保存不当、组织过少时,DNA 电泳可能无法看到明显条带。
A:推荐测序数据量至少为 6G。
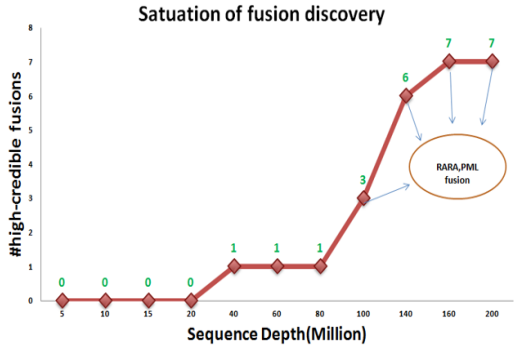
A:例如:Mycobacterium tuberculosis H37Rv。
Q:FPKM 的大小如何衡量?
A:一般认为 FPKM>1 是基因表达的,这个阈值是主流杂志推荐的;如果做 qPCR 验证,建议 FPKM>10。
A:用相关系数和 PCA,组内样本间的相关性应明显大于组间的相关性,才是比较成功的实验设计,差异基因会比较多。
A:(1)正面回答审稿人问题,不要企图回避;
(2)建议你把与结论重要的相关基因进行 qPCR 验证一遍,用数据说话:“虽然没有重复,但 qPCR 的结果支持测序结果;
(3)去数据库查找类似的研究数据来支持结论。